Wilson disease - update on pathophysiology and management
Dominik Huster, Karoline Leonhardt, Joachim Môssner
+ Pracoviště
Souhrn
Wilson disease is an autosomal recessive inherited disorder of human copper metabolism that leads to neurological symptoms and hepatic damage of variable degree. The affected gene, ATP7B, encodes a hepatic copper-transporting protein, which plays a key role in human copper metabolism. Clinical symptoms are divided in neurological symptoms such as tremor, dysarthria, psychiatric disorders etc.; predominant hepatic disease or mixed presentations. Copper deposition in the liver may result in acute liver failure, chronic hepatitis or liver cirrhosis. Early recognition by means of clinical, biochemical or genetic examination and early initiation of therapy with chelators or zinc salts are essential for favourable outcome and prognosis. Liver transplantation is an alternative in cases with acute and chronic liver failure and cures the hepatic disease. Frequent monitoring of drug therapy, adverse effects, and compliance is critical for the prognosis of the disease.
Key words: ATP7B - central nervous system -liver - copper – toxicity.
Copper as a trace element is essential for the human organism. Complex and precise interaction between several proteins is needed to maintain an appropriate copper balance in the cell. The liver plays a pivotal role in copper uptake, distribution and excretion in the human organism. The hepatic copper-transporting Wilson disease protein (ATP7B) plays a key role in the hepatic copper metabolism. Malfunction of ATP7B protein caused by ATP7B gene mutations leads to the Wilson disease, characterized by hepatotoxicity and neurological degeneration. In this review we summarize the current understanding of pathophysiology as well as diagnostic approaches, therapeutic strategies and surveillance of Wilson disease.
ASPECTS OF COPPER METABOLISM AND PATHOGENESIS OF WILSON DISEASE
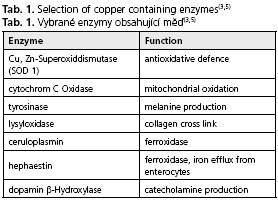
The average daily uptake of copper ranges between 1 and 2 mg, this amount is adequate for the needs of the human body. The majority of copper is taken up in the proximal parts of the small intestine(1-3). In the bloodstream copper is bound to amino acids (e.g. histidine) or proteins (e.g. albumin, transcuprin) and transported to the liver and peripheral tissues(4). Due to its redox potential copper is essential for many physiological processes in the cell and plays a critical role as a co-factor of important metabolic enzymes (Tab. 1)(3,5). A hereditary copper deficiency caused by mutations of the copper-transporting enzyme ATP7A, which is normally located in the gut and most of other tissues excluding the liver, leads to Menkes disease (X-chromosomal-linked) characterized by connective tissue disorders, severe neurodegeneration and early death(6).
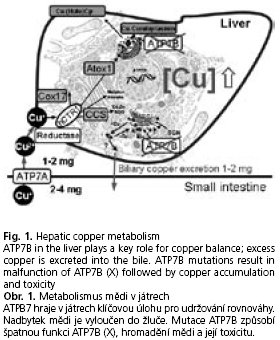
On the other hand again due to its redox properties cellular copper in its free or unprotected form affects cellular components such as membranes, enzymes or DNA. Therefore, several specific proteins, so-called chaperons, exist to protect the cell from deleterious effects of high copper reactivity(7). Copper chaperons bind and "escort" copper to miscellaneous cell compartments and enzymes(8). In case of cellular copper overload protection systems fail and elevated copper with its redox potential induces oxidative stress, which results in severe disturbances of cell integrity and function(9-11). Therefore, tight copper balance is indispensable. A major regulatory protein of copper homeostasis is ATP7B (Wilson disease protein), a copper-transporting Pj-type ATPase, mainly localized in the trans-Golgi network in hepatocytes(12). This large transmembrane protein transfers copper into the secretory pathway, mainly by binding it to ceruloplasmin. In case of excess cytosolic copper, ATP7B is responsible for biliary copper excretion(13) (Fig. 1).
Mutations in the ATP7B gene (autosomal recessive) cause various changes of the aminoacid sequence associated with alterations of protein structure and function, which lead to copper accumulation in the liver. This accumulation of copper results sooner or later in copper toxicity with liver disease of different severity and neurological and psychiatric disorders due to copper toxicity in the brain. The detailed reason and mechanism of copper toxicity in the brain is not fully understood. As a possible pathomechanism the release of large amounts of free copper into the blood stream followed by deposition of copper in neurons is discussed(4); however, experimental evidence for this assumption is still scarce.
INHERITANCE AND FREQUENCY
Kinnier Wilson firstly characterized and summarized the disease systematically in his thesis in 1912. Since 1948 the correlation between Wilson disease and copper overload is known (for overview see(14)). Beam described the autosomal recessive inheritance in 1953(15). The worldwide prevalence at birth is estimated to be approximately 1 in 30.000 with higher prevalences in certain populations. Thus, the gene frequency in the healthy population is about 1 in 90(16). Some authors also report a lower occurrence (1:40.000-1:75.000)(14-17). The gene ATP7B is located on the long arm of chromosome 13 (13ql4-q21). To date over 300 different mutations have been identified in this rather huge gene (cDNA: 4.4 kbp) (http://wwwmedicalgenetics.med.ualberta.ca/wilson/index.php)(18), whereas only few of these mutations are already functionally characterized(19-22). By far the most common mutation in Central, Eastern and Northern Europe is the point mutation H1069Q in exon 14, accounting for 30-60 % of all mutations in Caucasian patients(23-25). This mutation is associated with failure in catalytic phosphorylation and mislocalization of ATP7B(19,20). Other common mutations in Central and Eastern Europe are located on exon 8 (G710S, P767P-fs, W779X), exon 13 (R969Q), and exon 15 (P1134P-fs)(23-26). The allele frequency of most mutations is very low(23,25). A consequence of so many different mutations is that most patients carry two different mutations (so-called compound heterozygotes, e.g. M769V in combination with H1069Q)(4). Results of genetic studies in certain populations are not applicable for other regions and populations, therefore genetic testing is generally very complex and tedious.
CLINICAL MANIFESTATION OF WILSON DISEASE
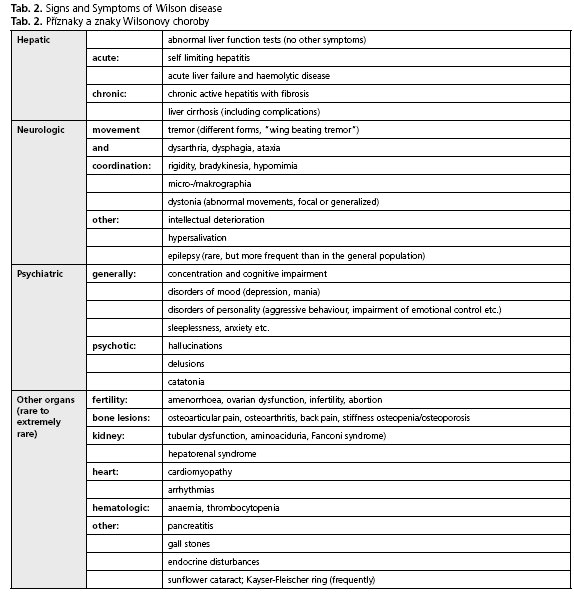
The Wilson disease shows a high level of heterogeneity with regard to symptoms and severity. In principle, the main features - the hepatic and the neurologic form - can be distinguished from one another, but many patients present with a mixture of both. Usually the hepatic form appears some years (between 8-18 years) prior to the neurologic form (early adulthood)(26-28). An overview of the most common symptoms is given in Table 2.
Liver disease
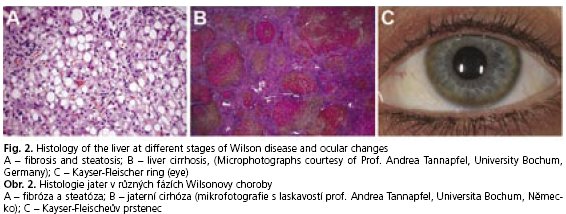
The clinical spectrum of liver disease in Wilson disease patients varies widely(29). It ranges from asymptomatic forms with elevated liver enzymes or liver enlargement, chronic hepatitis with fat accumulation, and fibrosis (Fig. 2A) to liver cirrhosis (Fig. 2B) and chronic liver failure. Unfortunately, there are also some individuals whose disease maybe obscured until their adolescent years, when they may present with acute fulminant liver failure. Normally it is associated with haemolytic anaemia (Coombs-negative), coagulopathy and acute renal failure(29). In some cases, however, it is difficult to distinguish between the histologie appearance of Wilson disease and chronic active hepatitis of different origins. Glycogen deposition in the nuclei, Mallory-bodies, and mitochondrial changes are typically seen, but these observations are rather unspecific(30).
Neurologic manifestation
The neurologic form is characterised by a cumulative motoric dysfunction. Initial symptoms are often unspecific, mild and may not be recognised. However, the most common presenting neurologic features include asymmetric distal accentuated tremor of the hands (postural tremor), "wing beating" tremor, intention tremor, and sometimes tremor of the trunk and shaking of the head. Such different kinds of tremor occur in approximately half of individuals with Wilson disease. Rigidity, dystonia and bradykinesia can be observed as well as dysarthria, dysphagia and difficulties with writing. Cerebral seizures occur rather rarely, but more frequent than in the general population. Dysreflexia, muscle weakness or loss of intelligence are not typical for Wilson disease (for detailed overview of neurologic signs see(14)).
Up to one third of the patients may suffer initially from psychiatric symptoms(14,28), including attention deficit disorders, depression, mood swings and in extreme cases psychotic symptoms. Psychiatric symptoms occur more frequently in patients with neurologic course, and it is important to mention that such symptoms have an organic basis and may dissolve in response to adequate therapy.
Granular deposition of elemental copper on the inner surface of the cornea (Descements membrane) causes the Kayser-Fleischer ring (Fig. 2C). The occurance of this phenomenon is an important diagnostic sign of the disease, but it is not associated with any loss of vision. Kayser-Fleischer rings are observed in up to 95 % of individuals with the neurologic form of Wilson disease, but in absence of neurologic symptoms only in 50-60 % of the cases(28).
DIAGNOSIS
Standard diagnostics
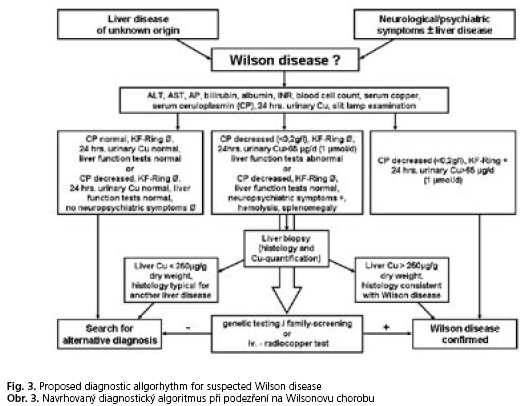
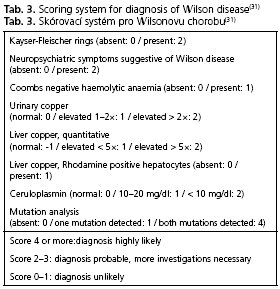
In case of clinical suspicion the diagnostic procedures for Wilson disease include mainly biochemical (in particular parameters of copper metabolism) and sometimes molecular biological investigations. The presence of typical neurologic signs or symptoms (Tab. 2), Kayser-Fleischer rings, low serum ceruloplasmin levels and elevated urinary copper excretion are sufficient to establish the diagnosis. Molecular genetic testing is important for testing presymptomatic siblings. In this case testing for hepatic copper concentrations is unnecessary. Nevertheless, follow-ups on liver function parameters are always very useful, because nearly all patients present with liver damages of various extent(1). To confirm the diagnosis a scoring-system (Tab. 3) can be helpful(31), but this has not yet been completely validated. In cases of obscure liver dysfunction work up includes copper metabolism parameters and, if necessary, a liver biopsy with histological evaluation and measurement of hepatic copper concentration. In some cases, additional genetic testing is needed to establish the diagnosis. A summary of the diagnostic steps in different clinical situations is shown in Figure 3.
Additional investigations
The intravenous radiocopper loading test is a reliable diagnostic tool in undefined cases(14). This method directly demonstrates failure of copper incorporation into ceruloplasmin and can even distinguish heterozygous carriers from patients with Wilson disease. Unfortunately the test is highly complex and is not routinely available.
The D-penicillamine challenge test may also be useful (especially in children), but it is not validated for adults and not a standard procedure(4).
An ultrasound scan of the liver is needed to identify and follow up liver damage and signs of liver cirrhosis(32,33).
Furthermore, a magnetic resonance imaging (MRI) of the brain appears to be more sensitive than CT scanning in detecting early cerebral changes and to monitor the course of Wilson disease. Thus, this method is gaining importance (see overview see(14)). Almost all patients with neurologic symptoms and many asymptomatic patients show lesions, especially in the basal ganglia. However, MRI alone is not sufficient to establish the diagnosis.
Genetics
Genetic studies are becoming more and more important for validation of the diagnosis and identification of affected family members who are still presymptomatic. Cox et al. have recently shown that the genetic diagnosis in asymptomatic patients is especially significant when biochemical parameters are inconclusive(27). Currently, molecular genetic studies include haplotype analyses, and direct mutation analyses through sequencing. In cases of known mutations limited sequencing after prior amplification of the region in question is used(29). It is important to mention that complete mutation analysis (in case of an unknown mutation) in the large gene ATP7B is still complex and rather expensive at present time.
THERAPEUTIC STRATEGIES
All Wilson disease patients - even in the presym-ptomatic stage - need lifelong drug therapy. In general pharmacological treatment alternatives include copper chelators and zinc salts. Essential for long-term success of pharmacological treatment for Wilson disease is the patients' adherence. Currently only liver transplantation is able to cure the disease (hepatic form). However, transplantation is only mandatory in some special situations (see below).
Drug therapy
Initial therapy in the clinical stage
It is assumed that symptomatic patients have an extensive copper overload, and the primary purpose of pharmacological treatment is a negative copper balance. This is accomplished by chelation therapy with D-penicillamine or trientine until the free serum copper level and urinary copper excretion is within the normal range. In most cases this lasts 6 to 12 months or even longer(17,29).
Maintenance therapy and therapy in presymptomatic stage
Maintenance therapy helps to keep copper levels in balance after previous copper reduction. Either low-dose chelators (compared to initial therapy) or zinc salts are used. Zinc salts block the intestinal absorption of dietary copper by stimulating the synthesis of a variety of endogenous copper chelators such as metallothioneins. This copper is excreted through desquamation of the gut epithelial cells within some days via faeces(34). Due to its relatively few side effects, zinc salts are highly suitable for therapy in the presymptomatic stage.
Therapy in special situations
Pregnancy: It can be assumed that in untreated Wilson disease patients, difficulties during pregnancy may occur. A discontinuation of the medication, for example, for fear of teratogenic effects of penicillamine, can lead to spontaneous abortion, severe relapses and even death from increasing copper toxicity. Pregnant patients should be advised to continue the medication throughout their pregnancy, because stopping drug therapy involves a high risk of disease progression or acute liver failure(29). The risks of the therapy for the unborn child are relatively low. In particular, zinc salts seem to be very save in reduced doses (25 mg TID)(17). However, only a few negative case reports about treatment with D-penicillamine during pregnancy have been published. Various case collections have shown numerous successful pregnancies without any fetal abnormalities(35). Trientine has teratogenic effects in animals; therefore it is advisable to use alternatives.
Acute liver failure: In cases of acute liver failure high effective chelation therapy with D-penicillamine is mandatory. A therapy with zinc salts alone is insufficient. In extreme circumstances orthotopic liver transplantation is indicated, when there is evidence of impending liver failure. Sometimes it is the only possible treatment option (see below). Until a graft becomes available, bridging management (e.g. albumin dialysis) can provide support(29).
Initial treatment of neurological symptoms: Initial chelator treatment of Wilson disease may impair neurological symptoms. A new drug for initial therapy of neurologic dysfunctions is tetrathio-molybdate, which is only used in the USA in context of clinical studies and has not been approved as Wilson disease therapy yet, although initial results are encouraging(36).
Surveillance
Drug therapy should be monitored relatively closely (biannual, at the beginning more frequently). Mentoring, observation of patient adherence as well as early detection of side effects, which may occur, are important for therapeutic success. Unfortunately, D-penicillamine can cause particularly serious, even irreversible side effects, which requires a change in therapy (such as nephrotic syndrome, anaemia, autoimmune disease). For providing evidence of the efficacy of zinc therapy an oral radiocopper test is used to show the successful blockade of intestinal copper absorption(37,38).
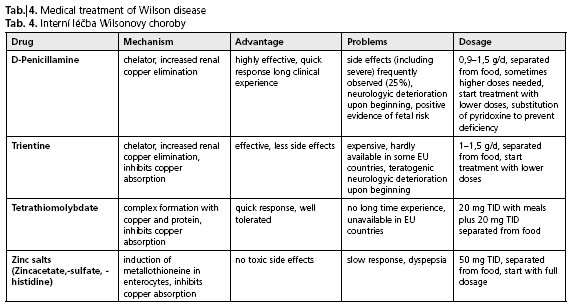
For more details, special side effects and further aspects of surveillance and monitoring the reader may refer to the wide range of specialist literature(14,17,29). The lack of clinical trials comparing different therapeutic strategies is a serious disadvantage for Wilson disease patients. Currently efforts on a multinational base in Europe have been started to improve this situation (for more information see http://www.eurowilson.org/). An overview of drug treatment of Wilson disease is provided in Table 4.
Liver transplantation
Transplantation of the liver is the only potentially curative treatment for Wilson disease. It is primarily indicated for treatment of patients with acute fulminant liver failure or end-stage liver cirrhosis, which progresses despite chelation therapy. Possible complications and the lifelong need of immuno-suppression are substantial risks, which should be taken into consideration. Several case reports have shown that a transplantation can improve neurologic symptoms, which otherwise seemed to be incurable(39,40). However, present data are not able to provide a general indication for liver transplantation to treat severe, therapy-resistant neurologic symptoms(28,29).
Adjuvant therapy
Application of antioxidants (e.g. vitamin E) is very useful to protect cells against oxidative damage of high copper concentrations(14). Because of the lack of evidence for efficacy and the lack of clinical trials validating its importance, antioxidants are not part of standard therapy of Wilson disease. Furthermore dietary modifications are helpful to support therapy, but as monotherapy diet alone is obsolete(29). The importance of a low copper diet as an adjuvant therapy is still debatable(14). Intake of foods rich in copper should be avoided, particularly during the initial phase of treatment. This applies to organ meats, chocolate, shellfish (especially lobster) and nuts. Complete abstinence under adequate medication is not necessary; but drinking water should not contain more than 2 mg of copper per litre according to the guidelines for drinking water quality and EU Directive. In households with water pipes made of copper alternatives such as mineral water are recommended.
Gene therapy
Gene therapy and/or stem cell therapies etc. might be an alternative curative therapy in the future. Results from animal studies using gene therapy and hepatocyte transplantation show that it is therapeutically effective, can reverse clinical symptoms and may have a good influence on the prognosis of the Wilson disease(41-43). However, such innovative approaches need further prospective developments and well defined safety rules to become an alternative therapy strategy.
PROGNOSIS/OUTCOME
If the disorder is detected early and treated properly (lifelong), then the patient suffering from Wilson disease may have a good prognosis including control of symptoms and normal life expectancy with an overall better outcome for patients with hepatic course(44). However, patients' adherence to treatment and frequent surveillance is essential for a successful therapy. Due to its rare incidence, variable progression with different symptoms and other possible problems we recommend caring for the patient with Wilson disease in specialized medical facilities in close cooperation with the general practitioner of the patient.Screening of family members
Siblings of Wilson disease patients have a 25% chance of having the disease, while children of patients have a 1 in 200 chance of developing the disease. All siblings should be tested; because the sooner the patients are diagnosed (even in presymptomatic stage) the better is their later outcome. In general, genetic screening of patients' children is not recommended, although they should receive regular medical check-ups(29). If the patient child presents abnormalities or precarious symptoms the diagnostic approaches should be intensified.
CONCLUSION
Undefined liver diseases and/or suspicious neurologic dysfunctions especially in young people are highly suggestive of Wilson disease. Particular attention should be given to an early and rather rapid diagnosis, because treatment delays may cause irreversible damages including long-lasting neurologic symptoms. A simple practical algorithm is useful to exclude or confirm the diagnosis of Wilson disease. The goals of treatment are reduction of the amount of copper in the tissues and maintenance of a low but adequate copper level in the organism.
Genetic testing is relevant for difficult cases or for screening of siblings. Consequent lifelong therapy, regular medical care and maximum therapy in critical situations (liver transplantation for treatment of patients with acute and chronic liver failure) have improved the prognosis enormously.
REFERENCES
- 1. Brewer GJ. Recognition, diagnosis, and management of Wilson's disease. Soc Biol 2000; 223: 39-46.
- 2. Schumann K, Classen HG, Dieter HH, et al. Hohen-heim consensus workshop: copper. Eur J Clin Nutr 2002; 56: 469-483.
- 3. Tapiero H, Townsend DM, Tew KD. Trace elements in human physiology and pathology. Copper. Biomed Pharmacother 2003; 57: 386-398.
- 4. Roberts EA, Cox DW. Wilson disease. Baillieres Clin Gastroenterol 1998; 12: 237-256.
- 5. Shim H, Harris ZL. Genetic defects in copper metabolism. J Nutr 2003; 133: 1527S-1531S.
- 6. Strausak D, Mercer JF, Dieter HH, et al. Copper in disorders with neurological symptoms: Alzheimer's, Menkes, and Wilson diseases. Brain Res.Bull. 2001; 55: 175-185.
- 7. Rae TD, Schmidt PJ, Pufahl RA, et al. Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science 1999; 284: 805-808.
- 8. Field LS, Luk E, Culotta VC. Copper chaperones: personal escorts for metal ions. J Bioenerg Biomembr 2002; 34: 373-379.
- 9. Gaetke LM, Chow CK. Copper toxicity, oxidative stress, and antioxidant nutrients. Toxicology 2003; 189: 147-163.
- 10. Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Curr Med Chem 2005; 12: 1161-1208.
- 11. Britton RS. Metal-induced hepatotoxicity Semin Liver Dis 1996; 16: 3-12.
- 12. Lutsenko S, Efremov RG, Tsivkovskii R, Walker JM. Human copper-transporting ATPase ATP7B (the Wilson's disease protein): biochemical properties and regulation. J Bioenerg Biomembr 2002; 34: 351-362.
- 13. Linder MC, Wooten L, Cerveza P, et al. Copper transport. Am J Clin Nutr 1998; 67: 965S-971S.
- 14. Hoogenraad TU. Wilson's Disease. Intermed Medical Publishers, 2001.
- 15. Bearn AG. Genetic and biochemical aspects of Wilson's disease. Am J Med 1953; 15: 442-449.
- 16. Schilsky ML. Wilson disease: genetic basis of copper toxicity and natural history. Semin.Liver Dis 1996; 16: 83-95.
- 17. Brewer GJ. Wilson's disease. A clinician's guide to recognicion, diagnosis, and management. Kluwer Academic Publishers, 2001.
- 18. Kenney SM, Cox DW. Sequence variation database for the Wilson disease copper transporter, ATP7B. Hum Mutat 2007; 28: 1171-1177.
- 19. Tsivkovskii R, Efremov RG, Lutsenko S. The role of the invariant Hisl069 in folding and function of the Wilson's disease protein, the human copper-transporting ATPase ATP7B. J Biol Chem 2003; 278: 13302-13308.
- 20. Huster D, Hoppert M, Lutsenko S, et al. Defective cellular localization of mutant ATP7B in Wilson's disease patients and hepatoma cell lines. Gastroenterology2003; 124: 335-345.
- 21. Forbes JR, Cox DW. Copper-dependent trafficking of wilson disease mutant ATP7B proteins. Hum Mol Genet 2000; 9: 1927-1935.
- 22. Forbes JR, Cox DW. Functional characterization of missense mutations in ATP7B: Wilson disease mutation or normal variant? Am J Hum Genet 1998; 63: 1663-1674.
- 23. Ferenci P. Regional distribution of mutations of the ATP7B gene in patients with Wilson disease: impact on genetic testing. Hum Genet 2006; 120: 151-159.
- 24. Polákova H, Katrincsakova B, Minarik G, et al. Detection of Hisl069Gln mutation in Wilson disease by bidirectional PCR amplification of specific alleles (BI-PASA) test. Gen Physiol Biophys ; 26: 91-96.
- 25. Vrabelova S, Letocha O, Borsky M, Kozák L. Mutation analysis of the ATP7B gene and genotype/phenotype correlation in patients with Wilson disease. Mol Genet Metab ; 86: 277-285.
- 26. Caca K, Ferenci P, Kuhn HJ, et al. High prevalence of the H1069Q mutation in East German patients with Wilson disease: rapid detection of mutations by limited sequencing and phenotype-genotype analysis. J Hepatol 2001; 35: 575-581.
- 27. Cox D, Prat L, Walshe JM, et al. The importance of molecular diagnosis for Wilson Disease. Hepatology 2003; 38: 230A.
- 28. Ferenci P. Pathophysiology and clinical features of Wilson disease. Metab Brain Dis 2004; 19: 229-239.
- 29. Roberts EA, Schilsky ML. A practice guideline on Wilson disease. Hepatology 2003; 37: 1475-1492.
- 30. Langner C, Denk H. Wilson disease. Virchows Arch 2004;445:111-118.
- 31. Ferenci P, CacaK, Loudianos G, et al. Diagnosis and phenotypic classification of Wilson disease. Liver 2003;23:139-142.
- 32. Vogel W, Kathrein H, Dietze O, Judmaier G. Ultra-sonography in Wilson disease. Ann Intern Med 1988; 108: 769-770.
- 33. Vogl T, Steiner S, Hahn D, Schedel H, Lissner J. Diffuse liver parenchymal diseases: the value of MRI compared to sonography and CT. Rofo 1991; 154: 495-503.
- 34. Sturniolo GC, Mestriner C, Irato P, et al. Zinc therapy increases duodenal concentrations of metallothionein and iron in Wilson's disease patients. Am J Gastroenterol 1999; 94: 334-338.
- 35. Sternlieb I. Wilsons disease and pregnancy (editorial; comment). Hepatology 2000; 31: 531-532.
- 36. Brewer GJ, Askari F, Lorincz MT, et al. Treatment of Wilson disease with ammonium tetrathiomolybdate: IV. Comparison of tetrathiomolybdate and trientine in a double-blind study of treatment of the neurologic presentation of Wilson disease. Arch Neurol 2006; 63: 521-527.
- 37. Siegemund R, Gunther K, Kuhn HJ, Lossner J. Medikamentose Beeinflussung der Bioverfugbarkeit von Kupfer. Zentralblatt fur Pharmakologie, Pharmako-therapie und Labordiagnostik ; 127: 239-240.
- 38. Hill GM, Brewer GJ, Juni JE, et al. Treatment of Wilson's disease with zinc. II. Validation of oral 64copper with copper balance. Am J Med Sci 1986; 292: 344-349.
- 39. Schumacher G, Platz KP, Mueller AR, et al. Liver transplantation: treatment of choice for hepatic and neurological manifestation of Wilson's disease. Clin Transplant 1997; 11: 217-224.
- 40. Geissler I, Heinemann K, Rohm S, et al. Liver transplantation for hepatic and neurological Wilson's disease. Transplant Proc 2003; 35: 1445-1446.
- 41. Irani AN, Malhi H, Slehria S, et al. S. Correction of liver disease following transplantation of normal rat hepatocytes into Long-Evans Cinnamon rats modeling Wilson's disease. Mol Ther 2001; 3: 302-309.
- 42. Ha-Hao D, Merle U, Hofmann C, et al. Chances and shortcomins of adenovirus-mediated ATP7B gene transfer in Wilson disease: proof of principle demonstrated in a pilot study with LEC rats. Z Gastroenterol 2002;40:209-216.
- 43. Merle U, Stremmel W, Encke J. Perspectives for gene therapy of Wilson disease. Curr Gene Ther 2007; 7: 217-220.
- 44. Merle U, Schaefer M, Ferenci P, Stremmel W. Clinical presentation, diagnosis and long-term outcome of Wilson disease - a cohort study. Gut 2007; 56: 115-120.
Pro přístup k článku se, prosím, registrujte.
Výhody pro předplatitele
Výhody pro přihlášené