Porucha polykání jako první projev desminopatie
Tereza Málková1, Miroslava Balaščaková2, Jana Zídková3, Jana Haberlová4, Hana Nestávalová5, Barbora Jírová1, Kristýna Zárubová1, Barbora Obermannová1
+ Pracoviště
Souhrn
Porucha polykání je závažný klinický příznak, který v dětském věku bývá součástí komplexního, často neurologicky podmíněného onemocnění. V níže uvedeném textu prezentujeme případ pacienta s velmi vzácnou příčinou dysfagie v kombinaci s velmi mírnými projevy kongenitální myopatie při geneticky podmíněné desminopatii s bialelickou homozygotní mutací v genu DES. Dominujícím projevem u našeho pacienta byla těžká forma poruchy polykání s masivními tichými aspiracemi tekuté i kašovité stravy, pro něž byl indikován k zavedení perkutánní endoskopické gastrostomie v kojeneckém věku. Kazuistika rozšiřuje fenotypové spektrum autozomálně recesivní desminopatie o manifestaci dysfagie jako iniciálního projevu a podtrhuje význam genetického přehodnocení u dětí s atypickou poruchou polykání.
Klíčová slova
porucha polykání, desminopatie, perkutánní endoskopická gastrostomie
Úvod
Desminopatie patří mezi vzácná, geneticky podmíněná onemocnění způsobená mutací v genu DES (OMIM: 125660) kódující desmin, protein tvořící klíčovou součást intermediárních filament svalového vlákna, která jsou nezbytná pro správnou svalovou strukturu a funkci. Vlivem mutací dochází ke změně exprese desminu a akumulaci patologických granulofilamentózních agregátů desminu ve svalové tkáni. V závislosti na typu a lokalizaci mutace je pozorována variabilní klinická manifestace i nástup příznaků zahrnující zejména progresivní svalovou slabost (typicky s kaudokraniální propagací) a kardiomyopatii až srdeční selhání [1,2].
V literatuře byly popsány jednotky s autozomálně dominantní a vzácně i autozomálně recesivní dědičností. V rodinách s autozomálně dominantní formou dědičnosti desminopatie převládá výskyt primomanifestace v mladé až střední dospělosti, zatímco u recesivní formy jsou pozorovány první příznaky v dětství až adolescenci a jejich fenotyp je výrazně variabilnější a závažnější [1,2].
Dosud byly popsány více než dvě desítky případů autozomálně recesivně dědičné desminopatie zahrnující homozygotní i složené heterozygotní mutace v genu DES. První příznaky u těchto pacientů dle kazuistických sdělení zahrnovaly kromě myopatie v oblasti dolních končetin a kardiálních projevů i svalovou slabost obličeje, krku a horních končetin, respirační selhání, synkopy či hypotonii [1,3,4].
Byly popsány vzácné případy s dysfagií jako iniciálním projevem či poruchou polykání v rámci progrese onemocnění. U skupiny pacientů s narušenou produkcí desminu byl pozorován unikátní fenotyp s mírnou formou myopatie překrývající se s projevy kongenitálního myastenického syndromu s rozvojem pletencové slabosti, ptózou, oftalmoplegií, fatickými a dysfagickými obtížemi [5–9].
Kazuistika
Představujeme nyní 2letého chlapce z fyziologické gravidity, narozeného ve 40. týdnu těhotenství akutní sekcí pro hrozící hypoxii plodu s Apgar skóre 3-10-10, somaticky bez nápadností, eutrofický. Ve 13. hod života zastižena epizoda apnoe s nutností stimulace, klinicky nápadné zahlenění a stridor. ORL nález bez známek laryngomalacie, výrazné množství hlenu v nosohltanu. Dále dominovaly dysfagické obtíže se spuštěním polykacího aktu, výživa byla zajištěna cestou nazogastrické sondy, kterou toleroval bez obtíží. Sonografie břicha a mozku s normálním nálezem, neurologicky bez výpadkové symptomatologie.
Po přeložení na naši kliniku jsme doplnili endoskopické vyšetření a videofluoroskopii polykacího aktu (FEES a VFS), kde byly přítomny masivní tiché postdeglutivní aspirace mléka z reziduí v hltanu při svalovém oslabení, na penetračně-aspirační škále odpovídající PAS skóre 8, netěsný velofaryngeální uzávěr (obr. 1).
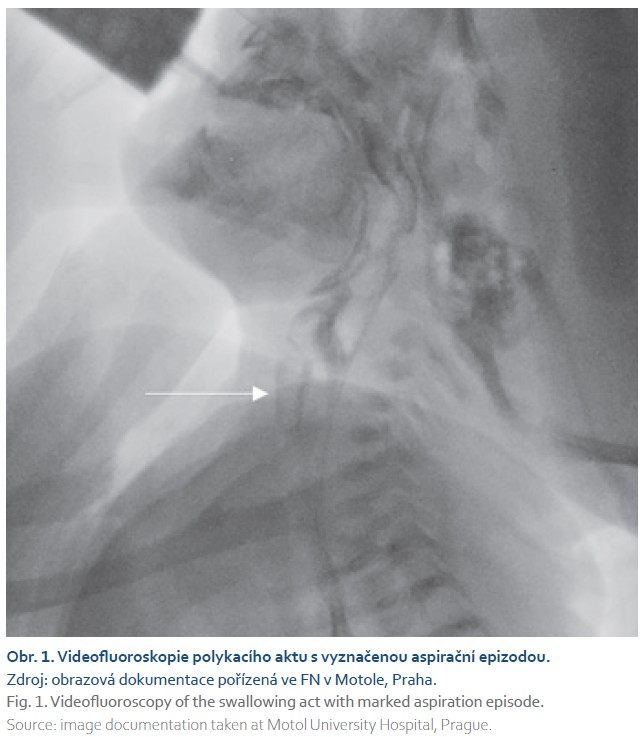
Magnetická rezonance mozku byla bez zásadních patologických změn, zahájili jsme genetické vyšetřování. Při rozvoji stolic s přítomností čerstvé krve a sonografickém zachycení plynu v portálním řečišti jsme vyslovili podezření na alergii na bílkovinu kravského mléka s přechodem na aminokyselinovou formuli podávanou cestou nazogastrické sondy. K nácviku polykání pod dohledem logopeda a k další péči byl chlapec přeložen do spádové nemocnice.
Další kontakt s pacientem v 6 měsících jeho věku, kdy přetrvávaly obtíže s polykáním tekutin, dle maminky příkrmy toleroval bez obtíží. Na kontrolní VFS byly opět přítomny tiché masivní aspirace tekutin, kaše i kaše s křupkami, které se nedaří navracet zpět do hltanu, odpovídající PAS skóre 8. Indikováno zavedení perkutánní endoskopické gastrostomie (PEG) a nastavena restrikční opatření při perorálním příjmu. Při klinickém vyšetření nově zachycena oboustranná ptóza a lehký konvergentní strabizmus, laboratorně přítomna elevace svalových parametrů kreatinkinázy v rozmezí 7–10 µkat/l a myoglobinu v rozmezí 90–160 µg/l. Z genetického vyšetření array CGH referována delece v oblasti 16p12.3 zasahující gen UMOD, který je obecně asociován s tubulointersticiálním onemocněním ledvin, avšak delece v souvislosti s tímto onemocněním nebyla dosud popsána. Nalezenou deleci jsme uzavřeli jako variantu nejasného významu (VUS), nesouvisející s fenotypovými obtížemi chlapce.
Při nově zjištěných nálezech a při podezření na kongenitální myopatii doplněno EMG, které bylo s normálním nálezem včetně repetitivní stimulace. Genetické vyšetření jsme rozšířili o panel genů asociovaných s neuromuskulárním onemocněním, kde byla detekována v genu DES pravděpodobně patogenní bialelická varianta c.1063C>T p. (Arg355*) v homozygotním stavu, která vede ke ztrátě funkce genu vlivem vytvoření předčasného terminačního kodonu. Kardiologické vyšetření bylo bez známek srdečního postižení. Kontrolní zhodnocení polykání v 1,5 roce s mírným zlepšením – PAS 6, aspirace jsou aktivně vyčištěny zpět do hltanu. Výživu zabezpečujeme cestou PEG, příkrmy matka podává v omezeném množství per os do rozvoje chrčení. Psychomotorický vývoj chlapce postupuje, od 1 roku obchází nábytek, od 16 měsíců samostatně chodí, ve 2 letech používá dvojslabičná slova s významem. Je v dobrém sociálním kontaktu, fixuje a sleduje bez omezení, přetrvává mírný konvergentní strabizmus a bilaterálně ptóza, mimika je slabší. Dominuje dysfagie a oslabení žvýkacích a polykacích svalů, trvá mírná hypotonie.
Diskuze
Nalezená varianta v genu DES c.1063C>T, p. (Arg355*) dosud nebyla v homozygotní formě popsána. Předpokládá se její patogenní účinek typu loss of function, který vzniká zařazením předčasného stop-kodonu s následnou trunkací proteinu. Rodiče pacienta jsou heterozygotní nosiči uvedené varianty, příbuzenský vztah negují a dosud nemají příznaky myopatie ani kardiální symptomatiky. U našeho pacienta byla tato varianta detekována ve věku 9 měsíců, kdy se ke svalové slabosti v oblasti hltanu s projevy dysfagie přidala lehká hypomimie s bilaterální ptózou. Z jednotek publikovaných případů autozomálně recesivně děděné desminopatie v homozygotní formě je zřejmé, že fenotyp je výrazně variabilní. Typicky se onemocnění projevuje svalovou slabostí nastupující během prvních dvou dekád života, avšak ne u všech pacientů se rozvine progresivní kardiomyopatie.
Náš pacient je v současnosti ve věku 2 let bez kardiosymptomatiky, nicméně zůstává sledován, neboť nelze vyloučit pozdější manifestaci kardiálních či dalších projevů onemocnění.
Závěr
Tímto kazuistickým sdělením bychom chtěli poukázat na důležitost multidisciplinárního přístupu u pacientů s poruchou polykání zahrnující spolupráci genetika, dětského neurologa, gastroenterologa i logopeda. Případ rozšiřuje fenotypové spektrum recesivních desminopatií o manifestaci těžké dysfagie již od novorozeneckého věku s velmi mírnými projevy kongenitální myopatie a zdůrazňuje potřebu dlouhodobého sledování nejen chlapce, ale i rodinných příslušníků a jejich edukaci stran možností prekoncepční diagnostiky.
ORCID autorů
M. Balaščaková 0000-0001-8827-4759,
J. Zídková 0000-0002-8729-6005,
J. Haberlová 0000-0003-2734-9715,
H. Nestávalová 0009-0002-4499-226X,
B. Jírová 0009-0000-8670-7434,
K. Zárubová 0000-0002-4720-0515,
B. Obermannová 0009-0002-5126-7349.
Doručeno/Submitted: 15. 11. 2025
Přijato/Accepted: 20. 11. 2025
Korespondenční autorka
MUDr. Tereza Málková
Pediatrická klinika
2. LF UK a FN v Motole
V Úvalu 84
150 06 Praha 5
Tereza.Malkova@fnmotol.cz
Pro přístup k článku se, prosím, registrujte.
Výhody pro předplatitele
Výhody pro přihlášené
Literatura
1. Goldfarb LG, Olivé M, Vicart P et al. Intermediate filament diseases: desminopathy. Adv Exp Med Biol 2008; 642: 131–164. doi: 10.1007/978-0-387-84847-1_11.
2. Clemen CS, Herrmann H, Strelkov SV et al. Desminopathies: pathology and mechanisms. Acta Neuropathol 2012; 125(1): 47–75. doi: 10.1007/s00401-012-1057-6.
3. Hauserman JG, Laverty CG, Donkervoort S et al. Clinical, immunohistochemical, and genetic characterization of splice-altering biallelic DES variants: therapeutic implications. HGG Adv 2024; 5(2): 100274. doi: 10.1016/j.xhgg.2024.100274.
4. Onore ME, Savarese M, Picillo E et al. Bi-allelic DES gene variants causing autosomal recessive myofibrillar myopathies affecting both skeletal muscles and cardiac function. Int J Mol Sci 2022; 23(24): 15906. doi: 10.3390/ijms232415906.
5. Goldfarb LG, Dalakas MC. Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease. J Clin Invest 2009; 119(7): 1806–1813. doi: 10.1172/jci38027.
6. Shah F, Franklin KA, Holmlund T et al. Desmin and dystrophin abnormalities in upper airway muscles of snorers and patients with sleep apnea. Respir Res 2019; 20(1): 31. doi: 10.1186/s12931-019-0999-9.
7. Polavarapu K, O’Neil D, Thompson R et al. Partial loss of desmin expression due to a leaky splice site variant in the human DES gene is associated with neuromuscular transmission defects. Neuromuscul Disord 2024; 39: 10–18. doi: 10.1016/j.nmd.2024.03.011.
8. Durmus H Ayhan Ö, Cirak S et al. Neuromuscular endplate pathology in recessive desminopathies: lessons from man and mice. Neurology 2016; 87(8): 799–805. doi: 10.1212/wnl.0000000000003004.
9. Cetin N, Balci-Hayta B, Gundesli H et al. A novel desmin mutation leading to autosomal recessive limb-girdle muscular dystrophy: distinct histopathological outcomes compared with desminopathies. J Med Genet 2013; 50(7): 437–443. doi: 10.1136/jmedgenet-2012-101487.