Kongenitální extrahepatální portosystémový shunt (Abernethyho malformace) komplikovaný rozvojem mnohočetného hepatocelulárního karcinomu – popis dvou případů
Irena Míková1, Jana Jarošová1, Darina Cupalová2, Andrea Vajsová3, Ondřej Fabián3,4, Eva Sticová Orcid.org 5, Tomáš Hucl Orcid.org 1, Libor Janoušek Orcid.org 6, Jiří Froněk Orcid.org 7, Pavel Taimr Orcid.org 1
+ Pracoviště
Souhrn
Úvod: Kongenitální extrahepatální portosystémový shunt (CEPS; Abernethyho malformace) může být komplikován rozvojem jaterní encefalopatie, plicní hypertenze nebo hepatocelulárního karcinomu (HCC). Prezentujeme případy dvou pacientů s mnohočetným HCC a současným CEPS. Popis případů: Dívka, 17 let, s mohutným portokaválním shuntem byla odeslaná na naše pracoviště pro progredující mnohočetná ložiska jater charakteru FNH/ adenomů dle MR. Cílená biopsie jednoho z ložisek prokázala hepatocelulární adenom s rizikovými histologickými znaky, nález byl chirurgicky neřešitelný a pacientka byla indikována k transplantaci jater (LT). V průběhu nekomplikované LT byl zrušen portokavální shunt. V explantátu jater byly přítomny mnohočetné adenomy jater, některé již s transformací do dobře diferencovaného HCC. Pacientka je 2,5 roku po LT s dobrou funkcí štěpu, bez recidivy HCC. Muž, 62 let, s mohutným portosystémovým shuntem mezi vena mesenterica superior a vena cava inferior a anamnézou jedné epizody jaterní encefalopatie byl odeslán na naše pracoviště pro CT nález dvou ložisek jater charakteru HCC v terénu pokročilé fibrózy jater (F3 dle elastografie). MR prokázala celkem šest ložisek jater velikosti do 25 mm charakteru HCC, AFP bylo 496 µg/ l. Nález překračoval indikační kritéria IKEM k LT pro HCC a pacient byl indikován k onkologické terapii. Závěr: Abernethyho malformace je spojena se zvýšeným rizikem rozvoje benigních (adenomy) a maligních tumorů (HCC) jater, pečlivá surveillance pacientů je nezbytná.
Klíčová slova
kongenitální extrahepatální portosystémový shunt, Abernethyho malformace, hepatocelulární karcinom, transplantace jater
Úvod
Kongenitální extrahepatální portosystémový shunt (CEPS) neboli Abernethyho malformace je vzácný stav, při němž většina splanchnické žilní krve obchází játra a je drenována přímo do systémové cirkulace skrz vrozený shunt mezi portální žílou (nebo jednou z jejích přívodných větví) a vena cava inferior (VCI) [1,2]. Poprvé byla tato malformace popsána Abernethym již v roce 1793 [3]. CEPS je často spojen s dalšími kongenitálními anomáliemi, nejčastěji kardiovaskulárními nebo urogenitálními, běžnou současnou abnormalitou je i polysplenie [1,2].
Ze zemí, kde se rutinně provádí neonatální screening hereditární galaktosemie, je odhad incidence kongenitálních portosystémových shuntů 1 : 30 000 narozených. V případě portosystémového shuntu galaktóza obchází játra a zvyšují se její sérové hladiny i při absenci enzymového deficitu. Galaktosemie však nedokáže odlišit extra- a intrahepatální shunty a nemusí navíc zachytit všechny případy CEPS [1,2]. Intrahepatální kongenitální portosystémový shunt (IPSS) je na rozdíl od CEPS častěji asymptomatický s možností spontánního uzávěru v raném dětství (do 2 let), nebyly u něj navíc popsány maligní tumory jater [1].
Někteří pacienti s CEPS mohou zůstat dlouhodobě asymptomatičtí, u jiných může dojít k rozvoji jaterní encefalopatie (HE), hepatopulmonálního syndromu (HPS) nebo plicní arteriální hypertenze (PaHT). Velmi časté jsou ložiskové léze jater včetně adenomů a hepatocelulárního karcinomu (HCC) [2].
Kongenitální extrahepatální portosystémový shunt existuje v mnoha variantách ve smyslu lokalizace, velikosti a postižených cév [2]. Na základě přítomnosti portálního toku v játrech se CEPS klasifikují na typ 1, což je end-to-side shunt, u něhož nejsou portální větve v játrech přítomné, a typ 2, kde je portální tok částečně odveden do systémové cirkulace v důsledku zachovalého nebo hypoplastického portálního kmene, který se side-to-side napojuje na VCI [1]. Ascites a portální hypertenze nejsou běžnou součástí CEPS na rozdíl od sekundárních portosystémových shuntů v rámci cirhózy nebo okluze portální žíly [2].
Problematika pacientů s CEPS byla shrnuta v multicentrické observační retrospektivní studii, která zahrnula 66 pacientů (53 % mužů) bez současné cirhózy [1]. Medián věku v době diagnózy byl 21 let (rozmezí 0– 66 let), medián sledování byl 5,2 let (0– 22 let). CEPS byl nejčastěji náhodně diagnostikován pomocí CT a/ nebo MR vyšetření. U 59 % pacientů se jednalo o typ 1, u 41 % o typ 2. U 29 % byla k dispozici biopsie jater, mezi typické znaky patřila absence nebo hypoplazie portálních venul v portálním poli kombinovaná v některých případech s hypertrofií arteriálních větví a kongescí sinusoid. Funkce jater byla zachovalá. U 44 % pacientů byla současně přítomna jiná malformace, nejčastěji kardiovaskulární. U 29 % pacientů došlo v průběhu sledování k rozvoji HE, převážně stupně I– II dle West-Haven kritérií, přičemž u většiny (84 %) se HE manifestovala před 25. rokem, u ostatních pak až po 50. roce věku. Sérové hladiny amoniaku v žilní krvi byly téměř u všech pacientů zvýšené bez ohledu na přítomnost HE a typ shuntu. U 65 % pacientů byla přítomna ložiska jater, která byla většinou mnohočetná. Z ložisek byly časté HCC (8×; medián věku 39 let, medián velikosti 94 mm), adenomy (10×; medián věku 18 let, medián velikosti 76 mm), fokální nodulární hyperplazie (FNH) (12×) a nodulární regenerativní hyperplazie (NRH) (7×). Mezi další komplikace CEPS patřily plicní arteriální hypertenze (PaHT) (8×) a hepatopulmonální syndrom (HPS) (2×).
Z důvodu adenomů/ HCC podstoupilo devět pacientů resekci jater a šest pacientů transplantaci jater (LT). U celkem 15 (23 %) pacientů byl proveden uzávěr shuntu v mediánu věku 17,5 let, a to z důvodu HE (4×), PaHT (3×), HPS (2×), velkého adenomu (1×) a preemptivně (5×). K uzávěru shuntu byla s ohledem na jeho velikost použita chirurgická ligace (11×) nebo perkutánní endovaskulární metoda (4×), přičemž v šesti případech byla použita dvoustupňová procedura s umožněním postupné redistribuce portálního toku do jater. U osmi pacientů s CEPS typu 1 byly po okluzi shuntu identifikovány intrahepatální portální větve, které dříve nebyly patrné. Uzávěr shuntu byl u jednoho pacienta komplikován rozsáhlou trombózou ve splanchnickém řečišti, u dalšího pacienta došlo v průběhu uzavírání shuntu k rozvoji významné portální hypertenze řešené zavedením TIPS (transjugulární intrahepatální portosystémový shunt), v ostatních případech byly výkon i následný průběh nekomplikované. V průběhu sledování zůstalo 32 % pacientů asymptomatických bez jakékoli intervence [1].
Popis případu 1
Dívka, 17 let, byla na naše pracoviště odeslána pro progredující mnohočetný ložiskový proces jater. V anamnéze měla defekt septa komor operovaný v novorozeneckém věku a primární amenoreu. Ve věku 13 let jí bylo po pádu z koně provedeno CT břicha s nálezem portokaválního shuntu (CEPS 1. typu) a vícečetných ložisek jater charakteru FNH. V roce 2020 byla na kontrolním MR jater v rámci pravidelné dispenzarizace zobrazena progrese velikosti a počtu ložisek, většina charakteru FNH, dvě z ložisek velikosti 46 mm a 72 mm charakterem odpovídala adenomu, u největšího ložiska byl možný i HCC. Cílená biopsie největšího ložiska na jiném pracovišti odpovídala FNH.
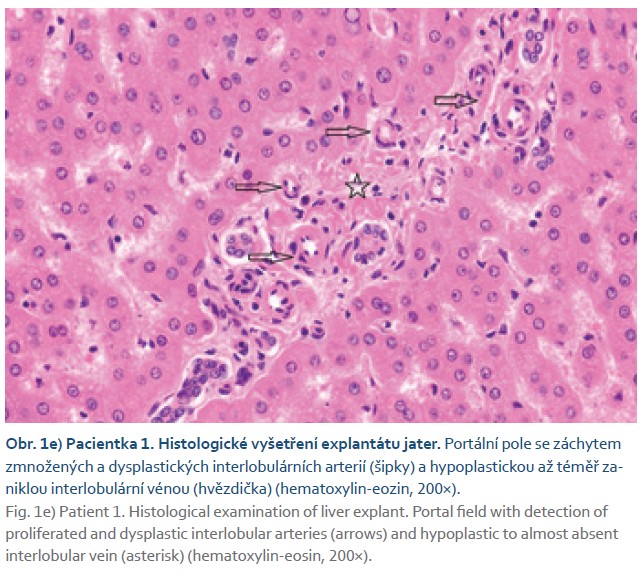
Vyšetření MR na našem pracovišti zobrazilo vena portae, která ústila do VCI, intrahepatální portální větve nebyly patrné (CEPS 1. typu) (obr. 1a). V játrech se zobrazila vícečetná ložiska o velikosti do 7 cm charakteru FNH-like lézí a dvě ložiska v pravém laloku jater odlišného charakteru, v.s. adenomy (obr. 1b). Cílená biopsie jednoho z těchto ložisek prokázala hepatocelulární adenom s mutací ß-kateninu, s ohledem na ložiskovou přítomnost některých rizikových histologických znaků (výpadky retikulinové sítě, pozitivita glypicanu 3 v některých hepatocytech) nebylo možno vyloučit agresivnější biologické chování. Tumor markery (AFP, CA 19-9, CEA) byly nízké. Na multidisciplinárním semináři byl nález zhodnocen jako inoperabilní a pacientka byla indikována k transplantaci jater (LT). V rámci předtransplantačního vyšetření a došetření námahové dušnosti byla zjištěna vrozená cirkulární stenóza průdušnice, která byla řešena kardiochirurgickou korekcí cévního prstence zužujícího tracheu. U pacientky byl dále diagnostikován syndrom polycystických ovarií. Ve věku 18 let u ní byla provedena LT, výkon byl chirurgicky nekomplikovaný, v průběhu LT byl zrušen vrozený portokavální shunt. V explantátu jater bylo přítomno celkem 12 ložisek v obou lalocích jater velikosti do 8 cm, histologicky adenomatóza jater (obr. 1c) s mutací ß-kateninu a v několika adenomech s transformací do dobře diferencovaného HCC (obr. 1d), několik z uzlů mělo charakter FNH-like lézí. V okolním jaterním parenchymu byly interlobulární vény v řadě polí hypoplastické nebo zaniklé, naopak interlobulární arterie byly na více místech zmnožené až s přítomností cévních konvolutů (obr. 1e). Pacientka je nyní 2,5 roku po LT bez obtíží, s dobře fungujícím štěpem jater, pravidelně menstruující, bez známek rekurence HCC.
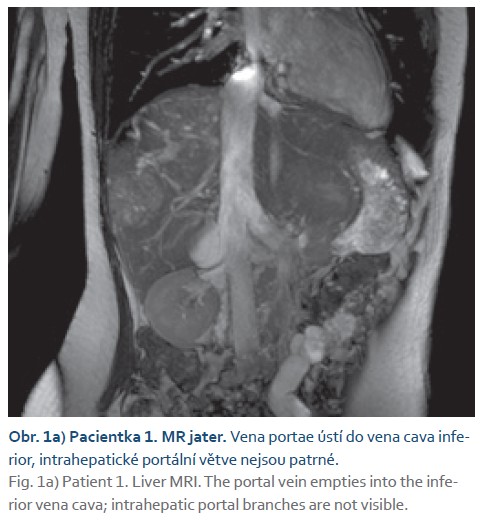
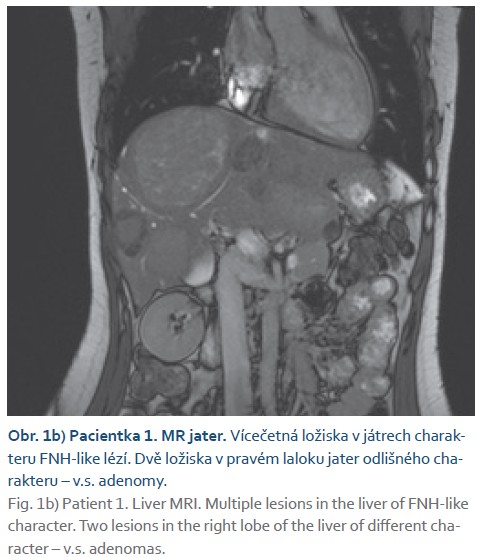
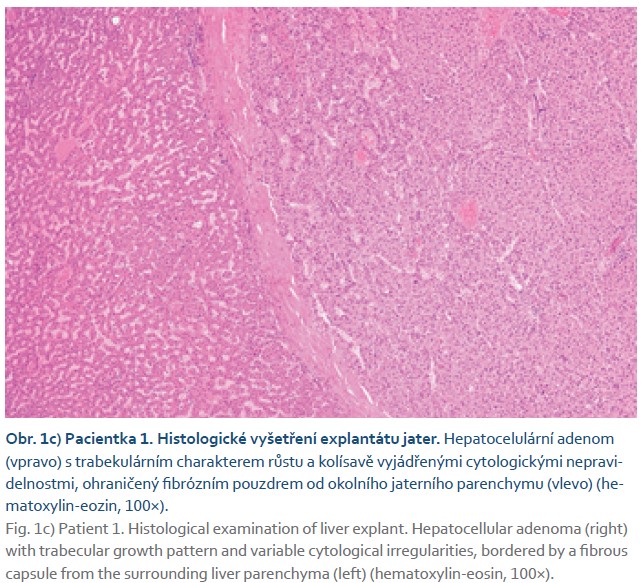
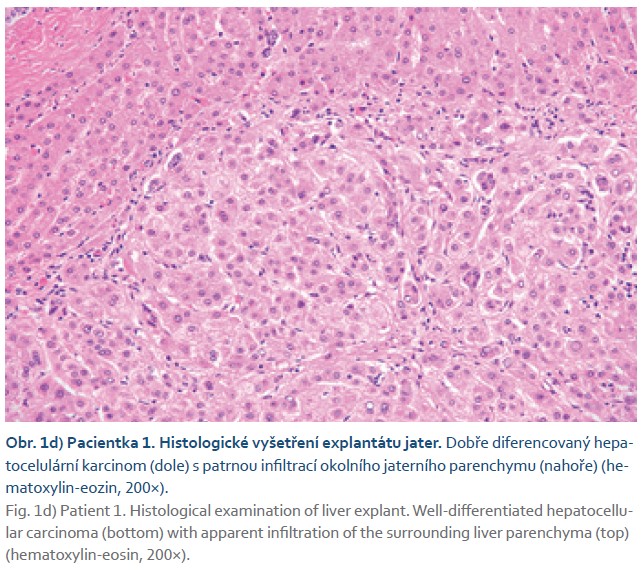
Popis případu 2
Muž, 62 let, s portosystémovým zkratem mezi v. mesenterica superior (VMS) a VCI, se dvěma ložisky jater charakteru HCC dle CT byl odeslán na naše pracoviště ke zvážení LT. V anamnéze měl diabetes mellitus 2. typu, fibrilací síní na antikoagulační léčbě, arteriální hypertenzi a obezitu. Pacient dlouhodobě konzumoval čtyři piva denně, v posledních šesti měsících abstinoval. Pro několik měsíců trvající únavu a malátnost byl hospitalizován v místě bydliště, kde byl stav hodnocen jako jaterní encefalopatie, která regredovala po terapii rixafiminem. Funkce jater byla v normě. Na CT břicha byl nalezen objemný portosystémový zkrat mezi VMS a VCI, dále byla v játrech zobrazena dvě ložiska charakteru HCC o velikosti 20– 25 mm.
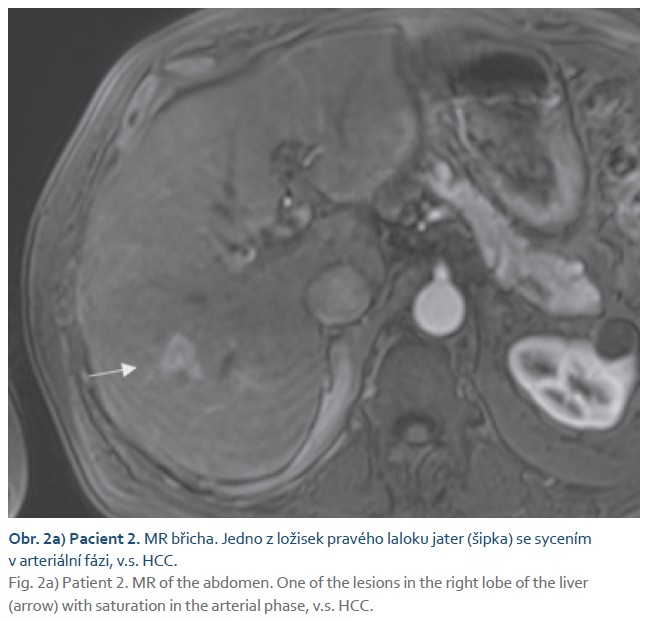
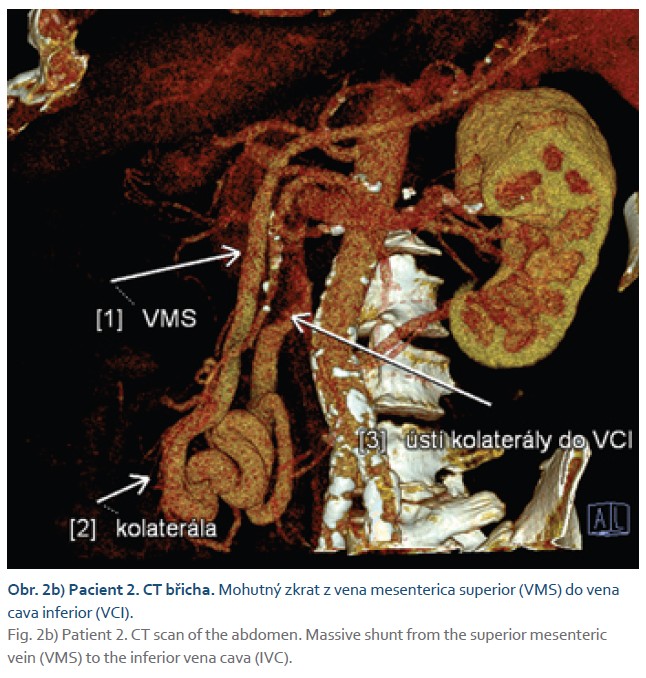
Dle MR jater na našem pracovišti bylo přítomno celkem šest ložisek jater velikosti do 25 mm, která měla všechna charakter HCC (obr. 2a). Játra měla dle MR charakter fibrózy či cirhózy s podílem steatózy, nebyly zde přítomny známky portální hypertenze (slezina 11 cm, bez ascitu, poměrně tenké vena portae, vena lienalis a VMS). Na CT břicha byl patrný mohutný zkrat z VMS do VCI (obr. 2b) a gracilní vena portae (typ 2 CEPS). Hodnota tuhosti jater 11,9 kPa dle shear wave elastografie odpovídala pokročilé fibróze (F3). Gastroskopicky nebyly přítomny varixy jícnu. Vzhledem k počtu ložisek a vysokému AFP (496 µg/ l) nálezy nesplnily Milánská ani rozšířená IKEM indikační kritéria k LT pro HCC a pacient byl indikován k onkologické léčbě na odesílající pracoviště.
Diskuze
V naší práci prezentujeme případy dvou pacientů s kongenitálním extrahepatálním portosystémovým shuntem (CEPS) neboli Abernethyho malformací, jejichž stav byl komplikován rozvojem mnohočetného HCC.
Kongenitální extrahepatální portosystémový shunt může zůstat dlouhodobě asymptomatický nebo se může v různém věku manifestovat jaterní encefalopatií, plicní arteriální hypertenzí (PaHT) nebo hepatopulmonálním syndromem (HPS). U velkého počtu (65 %) pacientů je CEPS komplikován ložiskovým procesem jater, který je často mnohočetný a může se vyskytnout v jakémkoli věku. V kohortě 66 pacientů s CEPS [1] došlo u 12 % pacientů k rozvoji HCC, a to již v mediánu věku 39 let, což je významně méně než průměrný věk pacientů s HCC u jiných jaterních chorob (73 let u MASLD, 66 let u HCV, 70 let u HBV) [4], u nichž je vznik HCC typicky vázán na rozvoj cirhózy jater. Mnohočetný HCC zjištěný v mladém věku jako u naší první pacientky tak u Abernethyho malformace není neobvyklý. HCC byl popsán již u 12měsíčního dítěte s CEPS [5].
Patofyziologii zvýšeného rizika jaterní malignity u CEPS lze vysvětlit významnou alterací lokální hemodynamiky jater. Přítomnost extrahepatálního portosystémového shuntu významně snižuje portální průtok játry a kompenzatorně se zvyšuje arteriální průtok jater. Snížený portální průtok a pokles jaterního metabolizmu splanchnických faktorů vede ke zvýšení cirkulujících hladin některých růstových faktorů, jako je inzulin, glukagon a růstový faktor hepatocytů, které mají stimulující účinky na játra. Maligní tumory u CEPS vznikají v absenci jaterní dysfunkce a cirhózy, a to buď de novo, nebo maligní transformací preexistujících benigních lézí. Celkově je riziko vzniku HCC u CEPS podobné jako u cirhózy a pacienti by měli mít pravidelnou surveillance. Ovšem i benigní tumory jater (adenomy, FNH) u CEPS mají jinou patogenezi a přirozený průběh a nejspíše nesou vyšší riziko maligního zvratu než běžné benigní tumory – doporučena je těsná surveillence pacientů. Podobně v patofyziologii PaHT a HPS hrají roli silné intestinální vazoaktivní mediátory, které obcházejí játra a přímo ovlivňují plicní řečiště [2].
U první prezentované pacientky byl portokavální shunt náhodně diagnostikován v mladém věku, pacientka měla přítomné další kongenitální kardiovaskulární malformace a neměla pokročilé chronické jaterní onemocnění, o vrozeném původu shuntu tak nebylo pochyb. U druhého prezentovaného pacienta však kongenitální původ shuntu nebyl zcela jednoznačný. Vyšší věk (66 let), obezita, dlouhodobá významná konzumace alkoholu (čtyři piva denně) a známky pokročilého jaterního onemocnění dle CT nás nutily pomýšlet na variantu sekundární portosystémové kolaterály v rámci cirhózy a portální hypertenze. Hodnocení tuhosti jater pomocí shear wave elastografie prokázalo stádium fibrózy F3 (průměr tuhosti jater 11,9 kPa), na CT břicha nebyly přítomny známky portální hypertenze (absence ascitu, splenomegalie a širšího portálního řečiště) ani nebyla přítomna žádná další portosystémová kolaterála. I u tohoto pacienta je tak pravděpodobný vrozený shunt, který byl dlouhodobě asymptomatický a manifestoval se až v pozdějším věku jaterní encefalopatií. Jaterní encefalopatie je častou (29 %) komplikací CEPS, přičemž se převážně jedná o encefalopatii stupně I– II dle West-Haven kritérií. Nejčastěji (84 %) se manifestuje již před 25. rokem, u menší části pacientů až po 50. roce věku [1].
Uzávěr shuntu chirurgickou nebo perkutánně endovaskulární cestou může vyřešit komplikace vyplývající z přítomnosti shuntu nebo zabránit jejich pozdějšímu vzniku, přičemž uzávěr shuntu je možný i u CEPS 1. typu, jelikož se po okluzi shuntu mohou intrahepatální portální větve otevřít [1]. S cílem umožnit postupnou redistribuci portální krve do jater bez vzniku komplikací může být uzávěr shuntu proveden jako dvoustupňový – nejdříve je zaveden stent redukující velikost shuntu následovaný definitivní okluzí po několika měsících [2]. U naší první pacientky byl vrozený shunt zrušen v průběhu LT, u druhého pacienta nebyl z důvodu rychlého vymizení HE po terapii rifaximimen a nálezu mnohočetného HCC uzávěr shuntu indikován pro předpokládaný malý klinický benefit. Uzávěr shuntu je dle literatury úspěšný zejména ve zlepšení HE, nižší účinnost byla popsána v případě dlouhodobé a těžké plicní arteriální hypertenze [1,6]. Uzávěr shuntu může vést k regresi počtu a/ nebo velikosti benigních tumorů jater [1,6] a lze jej v této indikaci rovněž doporučit. Při pochybnostech o benigní povaze ložisek, jako tomu bylo u naší první pacientky nebo v případě prokázané malignity, již klinický benefit uzávěru shuntu nelze očekávat a má být zvažována transplantace jater. Neinvazivní diagnostika ložiskových lézí u Abernethyho malformace a časná diagnostika HCC v terénu četných ložisek mohou být obtížné, o čemž svědčí celá řada nesrovnalostí v průběhu diagnostického procesu u naší první pacientky, odpovídají tomu i údaje z jiných kazuistických sdělení [7]. I v případě absence klinických symptomů bývá řadou autorů doporučována časná intervence k zabránění pozdějších komplikací [2], a to ideálně již při perzistenci shuntu po 2 letech věku [6].
Komplikace po uzávěru shuntu jsou dle dostupné literatury [1] jen málo četné. Uzávěr shuntu vedl k významné komplikaci pouze u dvou z patnácti pacientů, přičemž v jednom případě se jednalo o významnou portální hypertenzi řešenou založením TIPS a ve druhém případně o rozsáhlou mezenteriální trombózu [1].
Resekce nebo transplantace jater jsou metodou volby u HCC, indikovány bývají i u velkých adenomů jater. Oba námi prezentovaní pacienti byli zvažováni k LT z důvodu mnohočetného HCC nebo mnohočetných adenomů s rizikovými znaky. U mladé ženy s předoperační diagnózou mnohočetných adenomů byl až v explantátu jater prokázán mnohočetný, dobře diferencovaný HCC. U staršího muže přesahoval nález mnohočetného HCC s vysokým AFP Milánská i rozšířená IKEM indikační kritéria k LT a byla u něj indikována onkologická terapie.
Pro významné riziko benigních (adenomů) a maligních tumorů (HCC) jater je u pacientů s nálezem CEPS vhodný pravidelný screening ložiskových změn, racionální strategií může být podobně jako u cirhózy zobrazení jater každých 6 měsíců.
Závěr
Abernethyho malformace je spojena se zvýšeným rizikem rozvoje benigních a maligních tumorů (HCC) jater, surveillance ložiskových změn jater je nezbytná. Uzávěr shuntu chirurgickou nebo perkutánně endovaskulární cestou může vyřešit nebo zmírnit komplikace vyplývající z přítomnosti shuntu (jaterní encefalopatie, plicní arteriální hypertenze, hepatopulmonální syndrom) nebo zabránit jejich pozdějšímu vzniku.
ORCID autorů
I. Míková 0000-0001-7928-7558,
J. Jarošová 0000-0003-0010-0637,
D. Cupalová 0000-0002-6332-0461,
A. Vajsová 0000-0002-8473-8664,
O. Fabián 0000-0002-0393-2415,
E. Sticová 0000-0003-2486-6266,
T. Hucl 0000-0002-5648-4011,
L. Janoušek 0000-0001-7572-8416,
J. Froněk 0000-0003-2379-3886,
P. Taimr 0000-0002-6272-4608.
Doručeno/ Submitted: 16. 7. 2025
Přijato/ Accepted: 31. 7. 2025
Korespondenční autorka
MU Dr. Mgr. Irena Míková, Ph.D.
Klinika hepatogastroenterologie
Institut klinické a experimentální medicíny
Vídeňská 1958/ 9
140 21 Praha 4
irena.mikova@ikem.cz
Pro přístup k článku se, prosím, registrujte.
Výhody pro předplatitele
Výhody pro přihlášené
Literatura
1. Baiges A, Turon F, Simón-Talero M et al. Congenital extrahepatic portosystemic shunts (Abernethy malformation): an International Observational Study. Hepatology 2020; 71(2): 658– 669. doi: 10.1002/ hep.30817.
2. Papamichail M, Pizanias M, Heaton N. Congenital portosystemic venous shunt. Eur J Pediatr 2018; 177(3): 285– 294. doi: 10.1007/ s00431-017-3058-x.
3. Abernethy J. Account of two instances of uncommon formation in the viscera of the human body. Philos Trans R Soc Lond B Biol Sci 1793; 83: 295.
4. Huang DQ, El-Serag HB, Loomba R. Global epidemiology of NAFLD-related HCC: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 2021; 18(4): 223– 238. doi: 10.1038/ s41575-020-00381-6.
5. Benedict M, Rodriguez-Davalos M, Emre S et al. Congenital Extrahepatic Portosystemic Shunt (Abernethy Malformation Type Ib) With Associated Hepatocellular Carcinoma: Case Report and Literature Review. Pediatr Dev Pathol 2017; 20(4): 354– 362. doi: 10.1177/ 1093526616686458.
6. Franchi-Abella S, Branchereau S, Lambert V et al. Complications of Congenital Portosystemic Shunts in Children: Therapeutic Options and Outcomes. J Pediatr Gastroenterol Nutr 2010; 51(3): 322– 330. doi: 10.1097/ MPG.0b013e3181d9cb92.
7. Sharma R, Suddle A, Quaglia A et al. Congenital extrahepatic portosystemic shunt complicated by the development of hepatocellular carcinoma. Hepatobiliary Pancreat Dis Int 2015; 14(5): 552– 557. doi: 10.1016/ s1499-3872(15)60418-0.