Molekulární patogeneze karcinomu pankreatu
Tomáš Hucl Orcid.org 1, Julius Špičák Orcid.org 1
+ Pracoviště
Souhrn
Karcinom pankreatu představuje jednu z nejčastějších příčin úmrtí na malignitu u obou pohlaví. Snaha o pochopení molekulárně biologických procesů vedoucích postupně k jeho vzniku může tuto nepříznivou prognózu změnit, a» už umožněním včasné diagnostiky či cílené racionální terapie. Pravděpodobně více než desetina karcinomů vzniká v důsledku vrozené predispozice, za kterou jsou odpovědné geny BRCA2, CDKN2A, LKB1 či PRSS1. Stejně jako jiné epitelové karcinomy vzniká karcinom pankreatu postupnou progresí z neinvazivních prekurzorů. Tyto prekurzory, nazývané PanIN (Pancreatic Intraepithelial Neoplasia) společně s IPMN (Intraductal Papillary Mucinous Neoplasms) a MCN (Mucinous Cystic Neoplasms) tvoří skupinu lézí s maligním potenciálem, z nichž některé mohou být detekovatelné zobrazovacími metodami. Histologická progrese sleduje progresi genetickou. Vedle dobře známých mutací (K-ras, CDKN2A, p53, SMAD4) je dnes intenzivně studována role genů signální dráhy Fanconiho anémie. Znalost specifických genetických alterací je základem pro užití cílené racionální terapie. Souhrn identifikace pacientů s vrozenou predispozicí, včasné rozpoznání prekurzorů karcinomu pankreatu či nalezení specifických genetických či negenetických alterací umožňujících racionální terapii představují určitou naději na včasnou diagnostiku a účinnou léčbu tohoto onemocnění.
Klíčová slova: Fanconiho anémie - karcinom pankreatu - patogeneze.
ÚVOD
Adenokarcinomem pankreatu onemocní asi 100 osob na milion obyvatel za rok a přibližně stejný počet zemře. Jedná se o jednu z nejčastějších příčin úmrtí na malignitu u obou pohlaví. Karcinom pankreatu je většinou rozpoznán v jeho pokročilém biologickém stadiu. Pacient, nejčastěji mezi 60 a 70 lety, pozoruje vznik dyspepsie, váhový úbytek, bolest, změnu defekačních stereotypů nebo vznik ikteru či diabetu. Při vyšetření je nalezeno ložisko v pankreatu, většinou v jeho hlavě. U 80 % pacientů je zjištěn rozsáhlý případně generalizovaný nádor. Jedinou možností pro tyto pacienty je paliace (např. zavedení stentu do obturovaných žlučových cest) či nekurativní chemoterapie nebo chemoradioterapie. Pouze menšina nemocných je kandidátem chirurgické resekce, která jako jediná léčebná metoda prokazatelně prodlužuje přežití. I tito však nakonec většinou podlehnou generalizaci.
Narůstající poznatky o molekulární patogenezi karcinomu pankreatu představují naději na pochopení, a tím i účinnou diagnostiku a léčbu této choroby.
PREKURZORY KARCINOMU PANKREATU
Jako u jiných malignit nabízí prevence, vycházející ze znalosti rizikových faktorů, určitou naději na lepší prognózu. Věk, kouření, obezita, chronický zánět pankreatu, rasa, diabetes mellitus, dietní zvyky či přítomnost některých nádorových onemocnění v rodině riziko vzniku karcinomu významně zvyšují(1). Určitým způsobem prevence je také včasné rozpoznání anatomických lézí, které představují prekurzory adenokarcinomu pankreatu.
Drtivou většinu nádorů pankreatu představuje duktální adenokarcinom. Stejně jako jiné epitelové karcinomy i duktální adenokarcinom pankreatu vzniká postupnou progresí z neinvazivních prekurzorů. Již před sto lety byly popsány přechodné formy mezi normálním epitelem pankreatických vývodů a invazivním karcinomem jako buněčná hypertrofie, papilární formace a struktury nepravidelného růstu podobné karcinomu(2). Tyto prekurzory jsou dnes nazývány PanIN (Pancreatic Intraepithelial Neoplasia). Nejnižší stupeň, PanIN-1, je relativně častá a její výskyt se popisuje až u poloviny starých osob. Je odhadováno, že pouze 1 z 500 PanIN-1 lézí dospěje do stadia invaze. PanIN-1A je charakterizována náhradou kuboidních buněk pankreatických vývodů vysokými protáhlými buňkami s malými jádry lokalizovanými při bázi. V případě, že tyto buňky vytvoří papilární strukturu, hodnotí se jako PanIN-1B. PanIN-2 jsou méně časté, nalezené u asi 10 % starých osob, riziko malignizace je však větší. Jsou charakterizované abnormalitami jader, jako např. ztrátou polarity, zvětšením či kondenzací chromatinu. U PanIN-3 lézí nacházíme kribriformí shluky epiteliálních buněk oddělující se do lumen společně s nekrózami a těžšími jadernými atypiemi. PanIN-3 léze se většinou vyskytují společně s jednoznačným invazivním karcinomem(3). Progrese morfologická odpovídá s narůstajícími abnormalitami progresi genetické(4-7) (obr. 1).
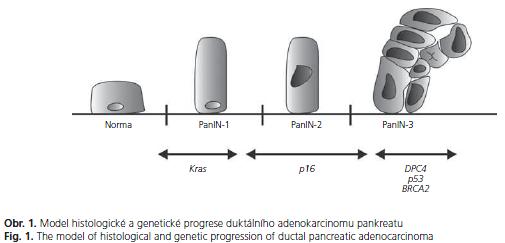
Jinou abnormalitou pankreatických vývodů s maligním potenciálem a možností vzniku duktálního adenokarcinomu jsou IPMN (Intraductal Papillary Mucinous Neoplasms). Jedná se o mucinózní nádory vycházející z hlavního či vedlejších pankreatických vývodů s různým stupněm dysplazie. Jejich průměrná velikost většinou nepřesahuje jeden centimetr a na rozdíl od PanIN lezí nejsou náhodným nálezem, ale mohou být detekovatelné zobrazovacími metodami. IPMN vycházející z hlavního pankreatického vývodu (main duct type) mají větší maligní potenciál a představují ve většině případů indikaci k chirurgickému řešení. Rozhodnutí o léčbě IPMN vycházející z větví hlavního vývodu (branch duct type) je složitější a záleží na více faktorech, kterými jsou přítomnost obtíží, modulární charakter a vztah k hlavnímu vývodu(8-10).
Mucinózní cystické nádory (MCN) nekomunikují s pankreatickými vývody, obsahují stroma ovariálního typu a vyskytující se především u žen. Většina z nich obsahuje adenomové struktury, v části případů je přítomen duktální adenokarcinom(8-10).
Přestože IPMN a MCN mohou dát vznik duktálnímu adenokarcinomu pankreatu, jejich genetické a epigenetické alterace se částečně liší(9) od klasického duktálního adenokarcinomu vzniklého bez jejich přítomnosti, ke kterému se vztahují údaje v následujícím textu.
GENETIKA
Adenokarcinom pankreatu je stejně jako jiná nádorová onemocnění genetickou a epigenetickou chorobou. V průběhu onkogeneze dochází postupně k akumulaci genetických a epigenetických alterací v onkogenech, tumor supresorových genech či genech udržujících neporušený genom. Somatické mutace v onkogenu KRAS2 se vyskytují u více než 95 % duktálních adenokarcinomů pankreatu, ale pouze u asi jedné třetiny medulárních karcinomů(11,12). CDKN2A gen (p16) je inaktivován u prakticky všech adenokarciomů mutacemi, homozygotními delecemi či utlumením transkripce metylací promotoru(13,14). Gen p53 je mutovaný u 50-75 %(15,16), gen MADH4 (DPC4, SMAD4) je inaktivovaný mutací či homozygotní delecí u asi 50 % nádorů(17). Somatické mutace mitochondriálního genomu jsou přítomny u téměř všech dobře studovaných případů(18).
Následující geny vykazují somatické mutace či homozygotní delece s nižší frekvencí: TGF-β receptory I a II (TGFBR1 a TGFBR2) (20), aktivinové receptory IB a II (ACVR1B a ACVR2) (21), LKB1/ STK11 kináza(22), RB1 substrát p16 regulovaných cyklin dependentních kináz(23), MKK4 mediátor stresem aktivované dráhy protein kináz(24), EP 300, BAX, BRAF, BUB1, FANCC, FANCG a BRCA2(25-29). Ve skutečnosti nese většina nádorů mutaci alespoň jednoho z těchto nízkofrekvenčních genů.
Většina adenokarcinomů pankreatu je charakterizována významnými strukturálními změnami chromosomů, jako jsou translokace, amplifikace, homozygotní delece či ztráty jednotlivých alel chromosomů, což vede k aneuploidii. Ztráty alel chromosomů (loss of heterozygosity) mají zásadní význam v tumorigenezi, nebo» umožňují manifestaci již přítomných heterozygotních mutací, které se tak stávají homozygotní. LOH postihuje u karcinomu pankreatu přibližně 40 % genomu se specifickou preferencí, především v oblastech tumor supresorových genů(19). Menšina adenokarcinomů je charakterizovaná normálním počtem chromosomů a mikrosatelitní instabilitou. Porucha opravy DNA vede k akumulaci mutací v repetitivních sekvencích genomu. V důsledku mikrosatelitní nestability dochází k charakteristickému postižení také v intragenových mikrosatelitech v kódujících frekvencích některých genů jako např. BAX, TGFBR2 či ACVR2(25,30).
Vedle mutací bylo popsáno několik míst genové amplifikace (AKT2, MYB, MDM2, KRAS2, AIB1(30-32).
MODEL HISTOLOGICKÉ A GENETICKÉ PROGRESE
Genetická progrese je provázena histologickou progresí charakterizovanou jednotlivými PanIN stadii. Téměř všechny PanIN-1 mají dramatické zkrácení telomer(33). Je možné, že toto poškození startuje chromosomální nestabilitu, která pak provází duktální tumorigenezi a konečný karcinom. Polovina PanIN-1 lézí má mutace v KRAS2 genu(34). Tato frekvence se zvyšuje u PanIN-2, kdy se také objevují p16 abnormality(35). Zvýšená exprese proteinu p53, která může signalizovat jeho mutaci, se objevuje u vyššího stadia PanIN. Ztráta Madh4 proteinu je charakteristická pro PanIN-3 léze(36) (obr. 1). Zdá se pravděpodobné, že tato uspořádaná progrese intraduktální neoplazie vede k takové konstelaci genetického poškození, které je nakonec odpovědné za invazivní a metastatický charakter tumoru(3,4).
VROZENÁ PREDISPOZICE
Patrně až jeden ze šesti karcinomů pankreatu vzniká na pozadí vrozené predispozice(37,38). Toto číslo je mezi karcinomy neobvykle vysoké. Pacienty s vrozenou dispozicí můžeme rozdělit do dvou skupin. První (asi 10 % všech karcinomů) je charakterizována nakupením rakoviny pankreatu v rodině. Toto nakupení může být součástí již známého vrozeného syndromu, většinou je však příčina vrozené dispozice neznámá. Její rozpoznání představuje jednu z největších výzev současné pankreatologie. V rodinách se dvěma či více postiženými členy má každý další člen rodiny 18násobné riziko vzniku karcinomu pankreatu. Rodiny se třemi členy mají přibližně 60násobně zvýšené riziko.
Druhou skupinou jsou rodiny, ve kterých výskyt karcinomu pankreatu budí dojem sporadického výskytu, přestože jejich členové jsou nositelé vrozených mutací genů asociovaných s nádory. Asi 7 % všech pacientů s karcinomem pankreatu je nositelem vrozené mutace v BRCA2 genu. Typický familiární způsob výskytu není u těchto pacientů většinou přítomen pro nízkou penetranci choroby u nositelů mutací(29).
Několik známých vrozených genetických syndromů představuje zvýšené riziko vzniku karcinomu pankreatu. Nejčastějším jsou BRCA2 mutace. Jak bylo zmíněno výše, mutace v BRCA2 genu typicky způsobují zdánlivě sporadické formy karcinomu díky jejich nízké penetranci. Vedle toho asi jedna ze šesti rodin s familiárním karcinomem s mnohočetným postižením členů (3 a více) je nositelem vrozené BRCA2 mutace(39).
Vrozené mutace genu CDKN2A způsobují FAMMM (Familial Atypical Multiple Mole and Melanoma) syndrom, spojený se zvýšeným rizikem výskytu melanomu a karcinomu pankreatu(40).
Vrozené mutace v genu LKB1/STK11 způsobují Peutz-Jeghers syndrom. Jsou přítomny hamartomové gastrointestinální polypy, melaninové skvrny sliznic a asi ve 30 % karcinom pankreatu(22,41).
Syndrom hereditárního nepolypózního kolorektálního karcinomu (HNPCC) způsobený vrozenými defekty v genech opravujících záměny v DNA také predisponuje ke karcinomu pankreatu. Tyto karcinomu jsou charakterizovány mikrosatelitní nestabilitou a mají často medulární fenotyp(42).
Vrozené mutace v PRSS1 genu způsobují hereditární pankreatitidu(43). Některé rodiny mají až 60násobné riziko vzniku karcinomu pankreatu(44). Předpokládá se, že karcinom vzniká jako důsledek zvýšeného obratu epiteliálních buněk duktů jako výsledek chronického zánětu. Toto představuje jedinečný příklad, kde vrozeně zvýšené riziko karcinomu není způsobené žádným ze tří základních typů tumorigenních genů.
NEGENETICKÉ ALTERACE
Nejen genetické alterace charakterizují karcinom pankreatu. Vedle například již zmíněného útlumu exprese genu CDKN2A pomocí hypermetylace byla popsána řada proteinů se zvýšenou expresí. Metoda SAGE (Serial Analysis of Gene Expression) například vedla k objevu PSCA (Prostate Stem Cell Antigen) či mesothelinu, membránových a secernovaných proteinů, které jsou zvýšeně produkovány většinou karcinomů pankreatu(45). Význam zvýšené exprese není vždy znám a je podrobně studován. Již nyní však víme, že představuje šanci k vytvoření nových metod detekce, zobrazení či terapeutického zacílení(46).
FANCONIHO ANÉMIE
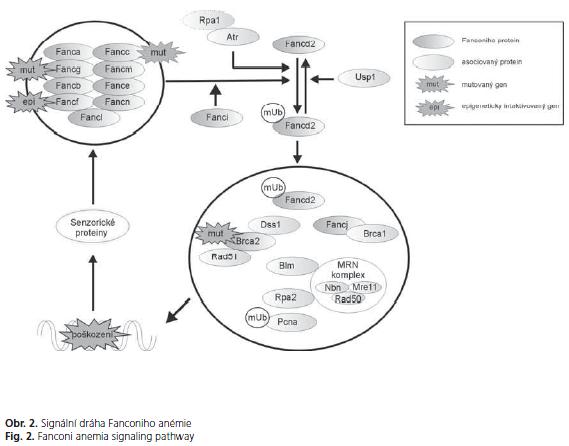
Genová rodina Fanconiho anémie (FA) se skládá z nejméně třinácti genů ( FANCA, FANCB, FANCC, BRCA2/FANCD1, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ, FANCL, FANCM, FANCN). Bialelické mutace v těchto genech způsobují FA, vzácnou recesivní chorobu charakterizovanou kongenitálními abnormalitami skeletu, progresivním selháním kostní dřeně a zvýšeným rizikem vzniku nádorů(47). Defekty v genech FA se vyskytují také u solidních nádorů v běžné populaci; FANCC a FANCG geny jsou mutované u karcinomů pankreatu(28) a epigenetický útlum FANCF je přítomen u řady nádorů(48). BRCA2/FANCD1 gen je mutovaný u pacientů s karcinomy pankreatu, prsu, ovarií a dalších orgánů (49).
Asi 7 % všech pacientů s karcinomem pankreatu je nositelem vrozené mutace v BRCA2/FANCD1 genu(29). Vedle toho asi jedna ze šesti rodin s familiárním karcinomem s mnohočetným postižením členů (3 a více) je nositelem vrozené BRCA2/FANCD1 mutace(39).
Krátce po zjištění, že gen BRCA2/FANCD1 patří do rodiny genů FA, byly nalezeny mutace v genech FANCC a FANCG u sporadických karcinomů pankreatu v nejméně 3 % procentech případů(28). Mutace byly nalezeny také mezi dostupnými buněčnými liniemi karcinomu pankreatu, což umožnilo získat předběžné výsledky o fenotypu nádorových buněk s poškozením signální dráhy FA(50).
FA proteiny účinkují ve společné signální dráze, jejíž distální část interaguje s regulátory buněčného cyklu a opravy DNA, především poškozené meziřetězovými můstky (interstrand-cross-links - ICL) a zlomením dvoušroubovice DNA (double strand break - DSB). Zformování základního nukleárního FA komplexu, skládajícího se z FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, FANCM a FANCN je závislé na integritě všech zúčastněných proteinů (obr. 2). FA komplex je aktivován po poškození DNA, které je detekované senzorickými proteiny jako ataxia telangiectasia mutated (ATM) nebo ataxia telangiectasia mutated and Rad3-related (ATR). Tato aktivace způsobuje monoubiquitinaci Fancd2, který následně přechází do jádra. Fancd2 kolokalizuje a interaguje s BRCA2 a mnoha dalšími proteiny účastnícími se opravy DNA jako BLM, BRCA1, NBN, PCNA, RAD51 a RPA2. Role BRCA2 po interakci s RAD51 spočívá především v opravě porušené DNA homologní rekombinací(51).
RACIONÁLNÍ TERAPIE
Hlavním důvodem studia molekulární patogeneze karcinomu pankreatu je snaha o jeho včasnější detekci a účinnější léčbu. Pojem „racionální terapie“ znamená, že znalost fundamentálních alterací určitého typu nádorového onemocnění umožňuje předpokládat úspěch určitých nových specifi ckých terapeutických přístupů. Například zvýšená exprese Her2/neu a EGFR (Epidermal Growth Factor Receptor) receptorů je předpokladem současně probíhajících studií užívajících monoklonální protilátky cílené proti receptorovým proteinům či studie užívající inhibitory receptorových kináz(52,53). Vysoký výskyt mutací KRAS2 u karcinomu pankreatu nabízel naději při užití farnesyl transferázových inhibitorů či jiných inibitorů funkce Ras proteinů. Téměř univerzální inaktivace Cdk inhibitoru, p16, slibovala úspěch při užití Cdk4 inhibitorů. Jakákoliv jiná relativní biochemická rozdílnost, která odliší rakovinové buňky karcinomu pankreatu, jako např. AKT2 nebo RelA aktivace, může představovat terapeutický cíl(25). Jednou z nevýhod tohoto přístupu, utilizujícího relativní rozdíly, je možný vývoj mutací v zamířeném genu, které způsobí rezistenci.
Alternativně lze využít absolutního biochemického rozdílu mezi nádorovými a nenádorovými buňkami. Tyto existují v místech genových delecí a inaktivujících mutací v nádorových buňkách. V tomto případě je cílem absolutní ztráta genové funkce, tudíž rakovinová buňka nemůže vyvinout rezistenci k léčbě. Inaktivované geny tak mohou být vhodné jako cíl nových terapeutik vysoce specifických pro nádorové buňky. BRCA2/FANCD1 mutace nabízí vhodný cíl na základě absolutního biochemického rozdílu. Buňky postrádající BRCA2 protein a buňky defektní v homologní rekombinaci poškozené DNA jsou ve srovnání s buňkami s intaktní signální dráhou FA zvýšeně citlivé k mitomycinu C či jiným preparátům způsobujícím meziřetězcové můstky nebo jiným látkám způsobujícím poškození DNA(50). Tato přecitlivělost může u pacientů s BRCA2 deficientními tumory vytvořit dostatečně široké terapeutické okno, potencionálně umožňující použití nízkých dávek chemoterapeutik s nízkou toxicitou po dlouhou dobu podávání(50,51). Nabízí se pak i možnost, že BRCA/FANCD1 status bude rutinním vyšetřením u pacientů s karcinomem pankreatu. Revoluční spekulací zůstává možnost profylaktické chemoterapie za účelem eliminace nádorových buněk se ztrátou wild-type (wt) allely u nosičů BRCA2 mutací, u kterých doposud karcinom nebyl diagnostikován.
Potenciální genově-specifická terapie karcinomů vznikajících na podkladě inaktivace FA proteinů bude vyžadovat vhodné preklinické modely. Všechny doposud vytvořené buněčné modely mají významné limitace pro farmakogenetické studie(51).
V reálné klinické medicíně se dnes můžeme setkat s několika příklady užití racionální terapie u karcinomu pankreatu, a» už v klinických studiích, či dokonce v běžné klinické praxi. Časté mutace v genu BRCA2, především v některých geografi ckých oblastech, ho činí v USA velmi atraktivním cílem. Probíhají klinické studie testující pacienty s nově diagnostikovanými karcinomy pankreatu na přítomnost BRCA2 mutací, u kterých je v případě pozitivity zahájena léčba mitomycinem C. Hodnotitelné výsledky nejsou ještě k dispozici (Scott E. Kern, osobní komunikace).
Výborná odpověď části nádorů plic na léčbu inhibitory tyrozin kináz vzbudila velké nadšení(52). To bylo o to větší, když se zdálo, že odpověď korespondovala s nálezem mutací v EGFR(53). Protože se tato specificita pro mutace v EGFR v plné míře nepotvrdila(54), nabízí se, že příčinou může být patologická aktivita v odpovídající signální dráze jiného původu. EGFR je zvýšeně exprimován či mutován u menšiny karcinomů pankreatu(55). Efekt EGFR inhibitorů byl studován také u pacientů s karcinomem pankreatu. Zdá se, že ve srovnání s gemcitabinem, mohou vést k mírnému zlepšení(56), je nutné však uvážit finanční náklady spojené s jejich užitím. Cílená terapie proti EGFR tak představuje jedinečný příklad složitosti problematiky racionální terapie. Po dlouhém laboratorním úsilí a detailním pochopení molekulárně-patogenetických mechanismů vyvinutá specifická terapie účinkuje, ne však stejně, jak se původně předpokládalo, a její podání způsobující u karcinomu pankreatu minimální benefit oproti stávající léčbě je zatížena obrovskými finančními náklady.
Selektivně nádorovými buňkami zvýšeně exprimované proteiny, jako je například mesothelin, mohou vyvolávat imunitní odpověď organismu. Ze studii Thomase et al. bylo patrné, že všichni pacienti, očkovaní stimulovanými autologními karcinomovými buňkami, kteří vyvinuli systémovou protinádorovou odpověď, měli silnou mesothelin specifickou CD8+ T-buňkami zprostředkovanou imunitní odpověď(57). Další vývoj těchto vakcín, stejně jako klinické studie užívající monoklonální protilátky proti mesothelinu, právě probíhají (Scott E. Kern, osobní komunikace). Racionalizace diagnostiky a terapie ve smyslu zohlednění nově objevených patogenetických principů jsou toužebně očekávaným výsledkem dlouhodobého úsilí mnoha výzkumných týmů. Vzhledem k omezeným výsledkům chirurgické či standardní onkologické léčby karcinomu pankreatu bez perspektivy další výrazné progrese představují nové patogenetické objevy jediný potenciál v léčbě tohoto závažného onemocnění.
LITERATURA
- 1. Gold EB. Epidemiology of and risk factors for pancreatic cancer. Surg Clin North Am 1995; 75: 819-843.
- 2. Hulst SPL. Zur kenntnis der Genese des Adenokarzinoms und Karzinoms des Pankreas. Trans. T. van Heek and V. Koopman. Vircdhows Arch B 1905; 180: 288-316.
- 3. Hruban RH, Adsaz NV, Albores-Saavedra J, et al. Pancreatic intraepithelial neoplasia: A new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol 2001; 25: 579-586.
- 4. Hruban RH, Maitra A, Kern SE, et al. Precursors to pancreatic cancer. Gastroenterol Clin North Am 2007; 36: 831-849.
- 5. Kern SE. Advances from genetic clues in pancreatic cancer. Curr Opin Onc 1998; 10: 74-80.
- 6. Hruban RH, Wilentz R, Kern SE. Genetic progression in the pancreatic ducts. Am J Pathol 2000; 156: 1821-1825.
- 7. Hruban RH, Adsay NV, Albores-Saavedra J, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol 2001; 25: 579-586.
- 8. Sohn TA, Yeo CJ, Cameron JL, et al. Intraductal papillary mucinous neoplasms of the pancreas: an updated experience. Ann Surg 2004; 239: 788-797.
- 9. Hruban RH, Maitra A, Kern SE, Goggins M. Precursors to pancreatic cancer. Gastroenterol Clin North Am 2007; 36: 831-849.
- 10. Bassi C, Sarr MG, Lillemoe KD, et al. Natural history of intraductal papillary mucinous neoplasms (IPMN): current evidence and implications for management. J Gastrointest Surg 2008; 12: 645-650.
- 11. Almoguera C, Shibata D, Forrester K, et al. Most human carcinomas of the exocrine pankreas contain mutant c-Kras genes. Cell 1988; 53: 549-554.
- 12. Wilentz RE, Goggins M, Redston M, et al. Genetic, immunohistochemical, and clinical feature sof medullary carcinomas of the pancreas: a newly described and characterized entity. Am J Pathol 2000; 156: 1641-1651.
- 13. Caldas C, Hahn SA, da Costa LT, et al. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet 1994; 8: 27-32.
- 14. Schutte M, Hruban RH, Geradts J, et al. Abrogation of the Rb/p16 tumor-supressive pathway in virtually all pancreatic carcinomas. Cancer Res 1997; 57: 3126-3130.
- 15. Pellegata S, Sessa F, Renault B, et al. K-ras and p53 gene mutations in pancreatic cancer: ductal and nonductal tumors progress through different genetic lesions. Cancer Res 1994; 54: 1556-1560.
- 16. Rozenblum E, Schutte M, Goggins M, et al. Tumorsupressive pathways in pancreatic carcinoma. Cancer Res 1997; 57: 1731-1734.
- 17. Hanh SA, Schutte M, Hoque AT, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996; 271: 350-353.
- 18. Jones JB, Song JJ, Hempen PM, et al. Detection of mitochondrial DNA mutations in pancreatic cancer offers a mass-ive advantage over detection of nuclear DNA mutations. Cancer Res 2001; 61: 1299-1304.
- 19. Hahn SA, Seymour AB, Hoque ATMS, et al. Allelotype of pancreatic adenocarcinoma using a xenograft model. Cancer Res 1995; 55: 4670-4675.
- 20. Goggins M, Shekher M, Turnacioglu K, et al. Genetic alterations of the TGF beta receptor genes in pancreatic and biliary adenocarcinomas. Cancer Res 1998; 58: 5329-5332.
- 21. Su GH, Bansal R, Montgomery E, et al. ACVR1B (ALK4) gene mutations in pancreatic carcinoma. Proc Natl Acad Sci USA 2001; 98: 3254-3257.
- 22. Su GH, Hruban RH, Bansal RK, et al. Germline and static mutations of the STK11/LKB1 Peutz- Jeghers gene in pancreatic and biliary cancers. Am J Pathol 1999; 154: 1835-1840.
- 23. Ruggeri B, Zhang SY, Caamano J, et al. Human pancreatic carcinomas and cell lines reveal frequent and multiple alterations in the p53 and RB-1 tumorsupressor genes. Oncogene 1992; 7: 1503-1511.
- 24. Su GH, Hilgers W, Shekher M, et al. Alterations in pancreatic, biliary, and breast carcinomas support MKK4 as a genetically targeted tumor-supressor gene. Cancer Res 1998; 58: 2339-2342.
- 25. Kern SE, Hruban RH, Hidalgo M, et al. An introduction to pancreatic adenocarcinoma genetics, pathology and therapy. Cancer Biol Ther 2002; 1: 607-613.
- 26. Calhoun ES, Jones JB, Ashfaq R, et al. BRAF and FBXW7 (CDC4, FBW7, AGO, SEL10) mutations in distinct subsets of pancreatic cancer: potential therapeutic targets. Am J Pathol 2003; 163: 1255-1260.
- 27. Hempen PM, Kurpad H, Calhoun ES, et al. A double missense variation of the BUB1 gene and a defective mitotic spindle checkpoint in the pancreatic cancer cell line Hs766T. Hum Mutat 2003; 21: 445.
- 28. van der Heijden MS, Yeo CJ, Hruban RH, et al. Fanconi anemia gene mutations in young-onset pancreatic cancer. Cancer Res 2003; 15: 2585-2588.
- 29. Goggins M, Schutte M, Lu J, et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res 1996; 56: 5360-5364.
- 30. Maitra A, Kern SE, Hruban RH. Molecular pathogenesis of pancreatic cancer. Best Pract Res clin Gastroenterol 2006; 20: 211-226.
- 31. Wallrapp C, Muller-Pillasch F, Solinas-Toldo S, et al. Characterization of a high copy number amplification at 6q24 in pancreatic cancer identifies c-MYB as a candidate oncogene. Cancer Res 1997; 57: 3135-3139.
- 32. Ruggeri BA, Huang L, Wood M, et al. Amplifi cation and overexpression of the AKT2 oncogene in a subset of human pancreatic ductal adenocarcinomas. Mol Carcinog 1998; 21: 81-86.
- 33. Van Heek NT, Meeker AK, Kern SE, et al. Telomere shortening is nearly universal in pancreatic intraepithelial neoplasia. Am J Pathol 2002; 161: 1541-1547.
- 34. Caldas C, Hahn SA, Hruban RH, et al. Detection of K-ras mutations in the stool of patiens with pancreatic adenocarcinoma and pancreatic ductal hyperplasia. Cancer Res 1994; 54: 3568-3573.
- 35. Moskaluk CA, Hruban RH, Kern SE. p16 and K-ras mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res 1997; 57: 2140-2143.
- 36. Wilentz RE, Iacobuzio-Donahue CA, Argani P, et al. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res 2000; 60: 2002-2005.
- 37. Lynch HT, Smyrk T, Kern SE, et al. Familial pancreatic cancer: a review. Semin Oncol 1996; 23: 251-275.
- 38. Klein AP, Hruban RH, Brune KA, et al. Familial pancreatic cancer. Cancer J 2001; 7: 266-273.
- 39. Murphy KM, Brune KA, Griffin C, et al. Evaluation of candidate genes MAP2K4, MADH4, ACVR1B, and BRCA2 in familial pancreatic cancer: deleterious BRCA2 mutations in 17%. Cancer Res 2002; 62: 3789-3793.
- 40. Goldstein AM, Fraser MC, Struewing JP, et al. Increased risk of pancreatic cancer in melanoma-prone kindreds with p16INK4 mutations. Ne Engl J Med 1995; 333: 970-974.
- 41. Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000; 119: 1447-1453.
- 42. Lynch HT, Voorhees GH, Lanspa SJ, et al. Pancreatic carcinoma and hereditary nonpolyposis colorectal cancer: a family study. Br J Cancer 1985: 52: 271- 273.
- 43. Whitcomb DC, Gorry MC, Preston RA, et al. Hereditary pankreatitis is caused by a station in the cationic trypsinogen gene. Nature Genet 1996; 14: 141-145.
- 44. Lowenfels AB, Maisonneuve P, DiMagno EP, et al. Hereditary pankreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. Journal of the National Cancer Institute 1997; 89: 442-446.
- 45. Ryu B, Jones J, Blades NJ, et al. Relationships and differentially expressed genes among pancreatic cancers examined by large-scale serial analysis of gene expression. Cancer Res 2002; 62: 819-826.
- 46. Hucl T, Brody JR, Gallmeier E, et al. High cancerspecific expression of mesothelin (MSLN) is attributable to an upstream enhancer containing a transcription enhacer factor dependent MCAT motif. Cancer Res 2007; 67: 9055-9065.
- 47. D‘Andrea AD, Grompe M. The Fanconi anaemia/ BRCA pathway. Nat Rev Cancer 2003; 3: 23-34.
- 48. Taniguchi T, Tischkowitz M, Ameziane N, et al. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med 2003; 9: 568-574.
- 49. Howlett NG, Tanigushi T, Olson S, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 2002; 297: 606-609.
- 50. van der Heijden MS, Brody JR, Dezentje DA, et al. In vivo therapeutic responses contingent on Fanconi anemia/BRCA2 status of the tumor. Clin Cancer Res 2005; 11: 7508-7515.
- 51. Gallmeier E, Kern SE. Targeting Fanconi anemia/ BRCA2 pathway defects in cancer: the signifi cance of preclinical pharmacogenomic models. Clin Cancer Res 2007; 13: 4-10.
- 52. Kris MG, Natall RB, Herbst RS, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA 2003; 290: 2149-2158.
- 53. Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004; 350: 2129-2139.
- 54. Cohen EE, Lingen MW, Martin LE. Response of some head and neck cancers to epidermal growth factor receptor tyrosine kinase inhibitors may be linked to mutation of ERBB2 rather than EGFR. Clin Cancer Res 2005; 11: 8105-8108.
- 55. Kwak EL, Jankowski J, Thayer SP. Epidermal growth factor receptor kinase domain mutations in esophageal and pancreatic adenocarcinomas. Clin Cancer Res 2006; 12: 4283-4287.
- 56. Moore MJ, Goldstein D, Hamm J, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group [NCIC-CTG]. J Clin Oncol 2007; 25: 1960-1966.
- 57. Thomas AM, Santarsiero LM, Lutz ER, et al. Mesothelin- specific CD8(+) T cell responses provide evidence of in vivo cross-priming by antigen-presenting cells in vaccinated pancreatic cancer patients. J Exp Med 2004; 200: 297-306.
Pro přístup k článku se, prosím, registrujte.
Výhody pro předplatitele
Výhody pro přihlášené