Vítejte na nových webových stránkách časopisu Gastroenterologie a hepatologie. Můžete zde nahrávat své články do redakčního systému a zároveň si prohlédnout bohatý archiv 80 ročníků článků časopisu. V případě potíží prosím napište na info@carecomm.cz
Aktuální číslo
Vol 80 No 2 (2026)
Publikováno dne 17. dubna 2026
Oznámení

Vychází 2. číslo roku 2026
Více…

Časopis Gastroenterologie a hepatologie – 2. díl roku 2026
Více…
Zveme vás na odborné akce roku 2026

Více…
Přehledová práce
Tomáš Nesnídal, Soňa Fraňková (Autor)
Světlana Adamcová Selčanová, Daniela Žilinčanová, Ľubomír Skladaný (Autor)
Původní práce
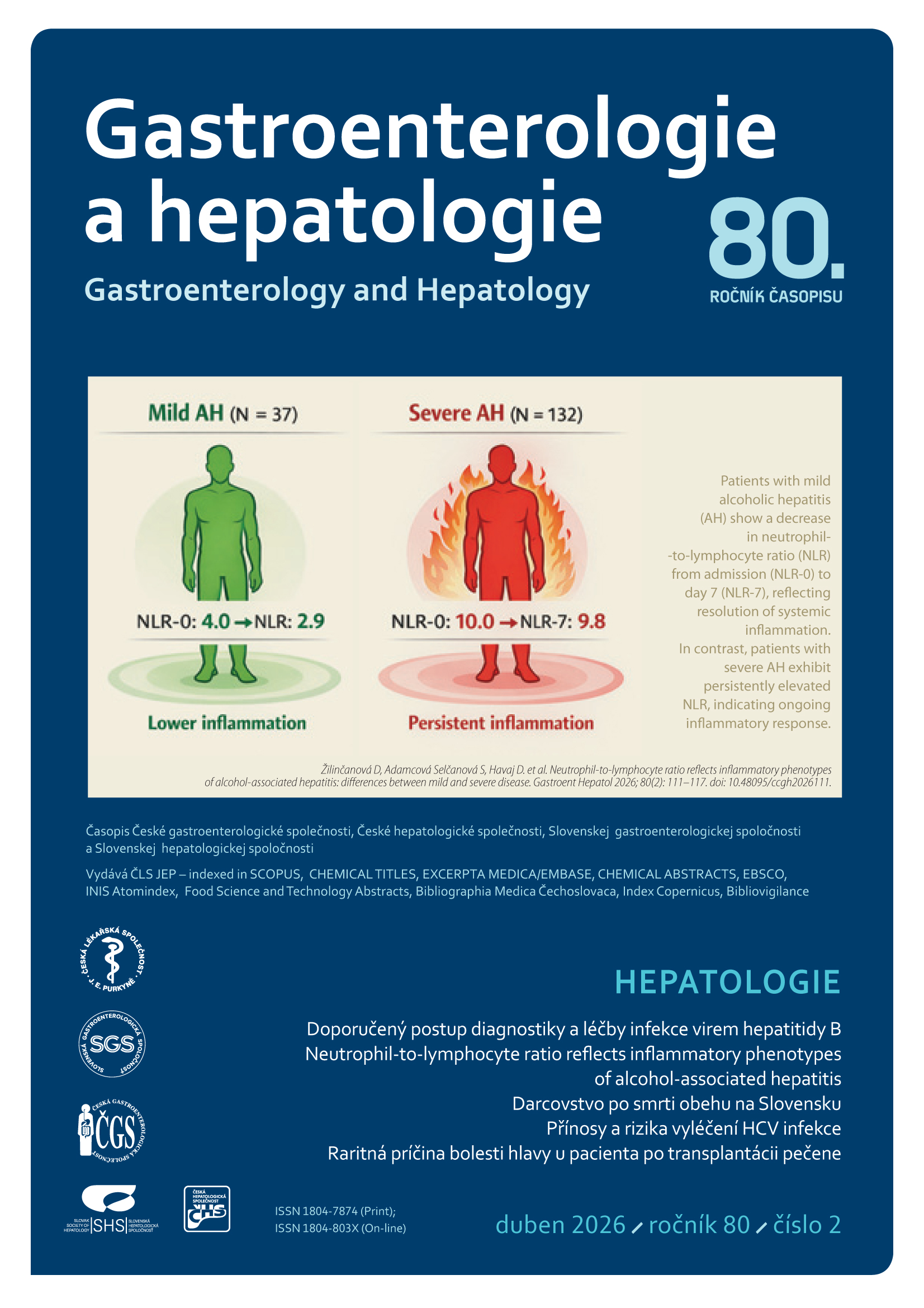
Daniela Žilinčanová, Světlana Adamcová Selčanová, Daniela Havaj, Karolína Šulejová, Natália Kubánek, Michaela Mihoková, Miroslav Výbošťok, Michal Žilinčan, Roman Záhorec, Peter Jarčuška, Ľubomír Skladaný (Autor)
Kazuistika
Natália Kubánek, Svetlana Adamcová Selčanová, Daniela Žilinčanová, Karolína Kristína Šulejová, Daniel Ján Havaj, Adam Halaj, Tomáš Koller, Ľubomír Skladaný (Autor)
Doporučené postupy
Petr Husa, Jan Šperl, Petr Urbánek, Soňa Fraňková (Autor)
Původní práce
Salikhat Dibraeva, Naida Iminova, Ilmuriyat Atasheva, Ravzanat Abdulgamidova, Rukizhat Aigunova, Karina Magomedova, Maryam Abakarova (Autor)
Přehledová práce
Petra Hlaušková, Karel Urbánek (Autor)
Komerčně podpořená sdělení
Michaela Bachratá, Jan Kulhavý (Autor)
Michaela Bachratá (Autor)