Wilsonova choroba v dětském věku – dvě kazuistiky
Nabil El-Lababidi1, Stanislav Houštěk2, Pavel Šochman3, Antonín Šípek4, Pavel Frühauf Orcid.org 5
+ Pracoviště
Souhrn
Dvě nepříbuzné dívky ve věku 5,5 a 8 let byly vyšetřeny pro náhodně zjištěné hepatopatie s hepatomegalií. Výsledky širokých laboratorních vyšetření sérologických, imunologických a metabolických vč. ceruloplazminu (CP) a odpadů mědi v moči byly bez patologií. Jaterní biopsie u obou dívek s histologickým nálezem jaterní steatózy a fibrózy různého stupně. Koncentrace mědi v jaterní sušině splňovala kritéria Wislonovy choroby (WD). Genetické vyšetření prokázalo u obou dívek přítomnost patogenní mutace genu ATP7B. Byla stanovena diagnóza WD a zahájená chelatační terapie D-penicilaminem s efektem. Toto kazuistické sdělení upozorňuje na skutečnost, že až 20 % dětských a dospělých pacientů s WD má fyziologické hodnoty CP a odpadů mědi v moči. Dále shrnuje nová doporučení Evropské společnosti pro dětskou gastroenterologii, hepatologii a výživu ohledně diagnostiky a léčby WD u dětí.
Klíčová slova
D-penicilamin, Wilsonova choroba, ceruloplazmin, gen ATP7B, měď, soli zinkuÚvod
Wilsonova choroba (WD – Wilson’s disease) je geneticky podmíněné onemocnění ovlivňující metabolizmus mědi [1]. Mutace při WD postihuje gen ATP7B, který kóduje transportní ATPázu typu P nutnou k vylučování mědi do žluči [2]. V důsledku této mutace dochází k progresivní toxické akumulaci mědi v játrech a jiných parenchymatózních orgánech. Tato akumulace začíná od zařazení potravin obsahujících měď do jídelníčku kojence. Při nerozpoznání a/nebo pozdním záchytu WD dochází k progresivnímu poškození jater a k rozvoji cirhózy a následnému jaternímu selhání. Předpokládaná prevalence WD je 1: 30 000 [1]. Nová data získaná molekulárním sekvenováním ukazují na mnohem vyšší prevalenci, až 1: 7 021 [3]. V dětském věku může WD probíhat jako asymptomatické jaterní onemocnění, jaterní cirhóza nebo akutní jaterní selhání. Neuropsychiatrické manifestace WD jsou v dětském věku výjimkou.
Následující dvě kazuistiky demonstrují záchyt asymptomatického průběhu WD u dvou nepříbuzných dívek.
Popis případů
Prvním pacientem je 8letá dívka, která byla od 7 let věku sledována pro náhodně zjištěnou smíšenou hepatopatii – alaninaminotransferáza (ALT) 2,56 μkat/l (0,05–0,60), aspartátaminotransferáza (AST) 1,46 μkat/l (0,10–0,63), gama-glutamyltransferáza (GMT) 0,83 μkat/l (0,10–0,39), alkalická fosfatáza (ALP) 8,6 μkat/l (1,12–6,20). Anamnéza dívky byla bezvýznamná. Klinicky byla hmatná játra přesahující žeberní oblouk o 1,5 cm, ostatní klinický nález byl chudý. Antropometrická data byla uspokojivá, výškou byla dívka na 45. percentilu a body mass index (BMI) na 86. Růstové křivky neprokazovaly propad ve sledovaných parametrech. Dívka byla široce vyšetřena: sérologie hepatotropních virů nesvědčila pro akutně nebo chronicky probíhající infekci, autoprotilátky byly negativní, široké metabolické vyšetření nesvědčilo pro event. vrozenou vadu metabolizmu, ceruloplazmin (CP) byl fyziologický, 0,3 g/l (0,2–0,6) a odpady mědi v moči nebyly zvýšené. Sonografie jater verifikovala hepatomegalii a echogenita jater byla difuzně zvýšená. Z důvodu diagnostických nejasností byla doplněna jaterní biopsie, kde byla histologicky hepatopatie charakterizována mírně periportálně akcentovanou smíšenou steatózou, hraničně balonovitě změněnými a ojedinělé zaniklými hepatocyty, absencí intraparenchymových neutrofilních granulocytů a mírnou periportální fibrózou. Měď v jaterní sušině byla enormně zvýšená (938 μg/g při normě < 250 μg/g). Genetické vyšetření verifikovalo přítomnost kauzální mutace p.His1069Gln genu ATP7B v homozygotní formě. U obou rodičů byla prokázána stejná mutace v heterozygotní formě. Byla stanovena diagnóza WD a nasazena chelatační terapie D-penicilaminem ve stoupajících dávkách s efektem. Jaterní testy se postupně normalizovaly, odpady mědi v moči po počátečním výrazném zvýšení postupně klesaly.
Druhým pacientem byla 5,5letá dívka s náhodně zjištěnou hepatopatií – ALT 3,12 μkat/l (0,05–0,60), AST 1,72 μkat/l (0,10–0,63), GMT 0,79 μkat/l (0,10–0,39), ALP 4,46 μkat/l (1,12–6,20) v rámci vyšetření 3 měsíce trvající únavy. Kromě únavy byla dívka zcela bez subjektivních stesků. Klinicky byla přítomna hmatná hepatomegalie přesahující oblouk žeberní o 2 cm. Dívka antropometricky dobře prospívala, výškou byla na 40. percentilu a BMI 77. Podobně jako v případě první dívky byly výsledky širokých vyšetření provedených k objasnění etiologie hepatopatie negativní nebo v rámci fyziologického rozmezí. CP (0,33 g/l) a odpady mědi v moči byly v normě. Ultrazvukové vyšetření jater verifikovalo hepatomegalii, jaterní parenchym byl s difuzně zvýšenou echogenitou a levý lalok jater zasahoval za horní pól sleziny. Byla provedena jaterní biopsie, kde byla histologicky těžká smíšená steatóza jater s výraznější kapénkovou složkou, periportální fibróza s incipientními přemostěními a mírná intralobulární fibróza. Měď v jaterní sušině byla zvýšená (503 μg/g). Bylo doplněno genetické vyšetření s nálezem dvou kauzálních patogenních mutací v genu ATP7B (p.His1069Gln a p.Gly710Ser) v heterozygotní formě. Byla stanovena diagnóza WD a byla zahájena chelatační terapie D-peniciliaminem s efektem. Postupně došlo k poklesu hodnot jaterních testů a ke zvýšení odpadů mědi v moči. Bylo doplněno genetické vyšetření u obou rodičů, které verifikovalo nosičství jedné patogenní mutace u každého z nich. Genetický screening WD u bratra dívky byl negativní.
Diskuze
Tradičně se uvádí, že WD je monogenně podmíněné, autozomálně recesivně děděné onemocnění [1,4]. Recentně byl dokumentován výskyt WD ve dvou nebo více po sobě jdoucích generacích. Tento výskyt svědčí pro tzv. pseudodominantní dědičnost [4,5]. Byly také dokumentovány případy atypické dědičnosti s výskytem tří mutací u jednoho pacienta a segmentální uniparenterální disomie [3,4]. WD nelze vyloučit jen na základě pozitivní rodinné anamnézy s výskytem onemocnění ve dvou a více po sobě jdoucích generacích. Dnes je identifikováno přes 600 patogenních variací genu ATP7B. Nejčastější je mutace p.H1069Q. Populační frekvence této alely je 10–40 % (30–70 % kavkazské populace). Mezi jiné časté mutace patří p.E1064A, p.R778L, p.G943S a p.M769V [4].
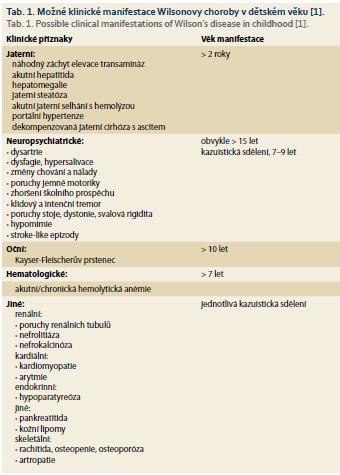
K akumulaci mědi v játrech a dalších parenchymových orgánech dochází od zavedení příkrmů s obsahem mědi. WD se může manifestovat kdykoliv od 3 do 74 let, v průměru ve 13,2 letech [1]. Klinické manifestace WD před 5. rokem věku jsou vzácné [1,6]. Projevy WD se dají rozdělit na jaterní, neuropsychiatrické, oční, hematologické a jiné (renální, kardiální, endokrinní a kostní) (tab. 1) [1]. Většina dětských pacientů se prezentuje s postižením jater. Rozsah postižení se pohybuje od náhodného záchytu elevace transamináz u dětí starších 1 roku přes akutní hepatitidu, hepatomegalii, sonografickou hyperechogenitu jater až po akutní jaterní selhání [1]. Neuropsychiatrické změny se manifestují ve druhém a třetím deceniu, méně často před 10. rokem života (4–6 % všech dětí s jaterním postižením jako první manifestací WD) [1]. Mírné poruchy kognitivních funkcí, zejména poruchy paměti a potíže s řečí, se zdají být relativně časté [7]. Kayser-Fleischerův prstenec obvykle není přítomen u dětí s asymptomatickým nebo primárně jaterním onemocněním. Tento prstenec je přítomen prakticky u všech dětí s neuropsychiatrickou symptomatikou [8]. Až v 6,9 % se může WD choroba manifestovat u dětí akutní hemolýzou, která je obvykle spuštěná infektem či léky a je přítomna zejména u fulminantních forem WD [9].
V diagnostice WD se tradičně užívá CP, který na sebe váže až 90 % volné mědi. Hladina CP je fyziologicky nízká u novorozenců a kojenců. Stanovení hladiny CP se nedoporučuje před dovršením 1 roku věku. Obvyklá hladina CP při WD je < 0,2 g/l. Tato hodnota má však nízkou prediktivní hodnotu nepřesahující 48,3 % [10]. Obdobný výsledek je u 20 % heterozygotních nosičů WD, při jaterním selhání, malabsorpci, poruchách glykosylace, Menkesově chorobě, malnutrici, nefrotickém syndromu, tzv. protein-losing enteropathy a při hereditární aceruloplazminemii [10]. Nejvyšší senzitivita (93 %) a specificita (100 %) byla nalezena při hodnotách CP < 0,14 mg/l [10]. Celkem 20 % dětí a dospělých s WD má však fyziologické hodnoty CP [1]. Dalším diagnostickým testem je měření odpadů mědi v moči. Za optimální cut-off hodnotu se považuje 0,65 μmol/24 hod se senzitivitou 78,9 % a specificitou 87,9 % [11]. Test s penciliaminem se považuje v diagnostice asymptomatických dětí za nespolehlivý a jeho standardní používání se nedoporučuje (senzitivita testu 12 %, specificita 47 %) [1,11]. V dnešní době je dostupná a rutinně používaná mutační analýza známých mutací v genu ATP7B. Nové sekvenční metody úspěšně identifikují obě mutované alely až v 95 % [1]. V nejednoznačných případech se doporučuje měření mědi v jaterní sušině. Za patognomické se považují hodnoty > 250 μg/g jaterní sušiny. S takovými hodnotami se setkáváme i u pacientů s primární biliární cirhózou, primární sklerozující cholangitidou a v některých případech poruch glykosylace [1]. Samostatné histologické vyšetření vzorku jaterní tkáně je nespecifické a nelze na jeho podkladě stanovit diagnózu WD [1,12]. V diagnostice WD pomáhá tzv. Ferenciho skóre, které hodnotí nejen klinické, ale i laboratorní parametry pacienta. Diagnóza se považuje za jistou při hodnotě ≥ 8 bodů.
Recentní doporučený diagnostický postup u dětí dle Evropské společnosti pro dětskou gastroenterologii, hepatologii a výživu z roku 2018 viz schéma 1.
Po stanovení diagnózy WD se doporučuje provedení screeningu tohoto onemocnění u příbuzných prvního stupně [1,13]. Tento screening slouží k záchytu asymptomatických pacientů a/nebo heterozygotních nosičů mutací WD. Ve screeningu se doporučuje provedení fyzikálního vyšetření, vyšetření jaterního souboru, stanovení hladiny CP a vyšetření identifikovaných mutací u pacienta [1]. Při screeningu dětí lze vyčkat do dosažení věku 1–2 let.
V terapii WD u dětí se, podobně jako u dospělých, užívá chelatační léčba D-penicilaminem nebo trientinem nebo blokace střevního vstřebávání mědi solemi zinku. Dietní omezení potravin bohatých na měď nevede k prevenci ukládání mědi v těle a doporučuje se nasazení diety do remise klinických a laboratorních příznaků onemocnění [1]. Z chelátů je v našich podmínkách k dispozici D-penicilamin, který je účinný u > 80 % symptomatických dětí [1]. Závažné nežádoucí reakce vedou k ukončení terapie až ve 30 %. Mezi nežádoucí reakce patří horečka, výsev vyrážky, neutropenie, trombocytopenie, lymfadenopatie a proteinurie [3]. Soli zinku se v dnešní době těší velké oblibě v terapii presymptomatických pacientů a jako udržovací terapie po vyplavení zásob mědi z těla chelátem. Monoterapie zinkem u symptomatických pacientů je velmi kontroverzní [14,15]. Klinické studie dokumentovaly lepší toleranci soli zinku než D-penicilaminu a lze je používat v terapii presymptomatických dětí [15]. Selhání terapie těmito solemi bylo prokázáno při terapii symptomatických dětí [14]. Zahájení terapie WD solemi zinku také představuje riziko zhoršení neuropsychiatrické symptomatologie [14].
Kolektiv autorů by rád poděkoval MUDr. Heleně Hůlkové a MUDr. Janu Stříteskému z Ústavu patologie 1. LF UK a VFN v Praze za histologické vyšetření vzorků jaterní biopsie.
Tento článek vznikl za podpory grantu Ministerstva zdravotnictví ČR RVO VFN 64165/2012.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
The Editorial Board declares that the manuscript met the ICMJE „uniform requirements“ for biomedical papers.
Doručeno/Submitted: 31. 8. 2018
Přijato/Accepted: 10. 10. 2018
MUDr. Nabil El-Lababidi
Centrum dětské gastroenterologie, hepatologie a výživy
Klinika dětského a dorostového lékařství 1. LF UK a VFN v Praze
Ke Karlovu 2
120 00 Praha 2
nabil.el-lababidi@vfn.cz

Pro přístup k článku se, prosím, registrujte.
Výhody pro předplatitele
Výhody pro přihlášené
Literatura
1. Socha P, Janczyk W, Dhawan A et al. Wilson’s disease in children: A position paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. J. Pediatr Gastroenterol Nutr 2018; 66 (2): 334–344. doi: 10.1097/MPG.0000 000000001787.
2. Bandmann O, Weiss KH, Kaler SG. Wilson’s disease and other neurological copper disorders. Lancet Neurol 2015; 14 (1): 103–113. doi: 10.1016/S1474-4422 (14) 70190-5.
3. Coffey AJ, Durkie M, Hague S et al. A genetic study of Wilson’s disease in the United Kingdom. Brain 2013; 136 (Pt5): 1476–1487. doi: 10.1093/brain/awt035.
4. Chang IJ, Hahn SH. The genetics of Wilson disease. Handb Clin Neurol 2017; 142: 19–34. doi: 10.1016/B978-0-444-63625-6.00003-3.
5. Dzieżyc K, Litwin T, Chabik G et al. Families with Wilson’s disease in subsequent generations: clinical and genetic analysis. Mov Disord 2014; 29 (14): 1828–1832. doi: 10.1002/mds.26 057.
6. Lin LJ, Wang DX, Ding NN et al. Comprehensive analysis on clinical features of Wilson’s disease: an experience over 28 years with 133 cases. Neurol Res 2014; 36 (2): 157–163. doi: 10.1179/1743132813Y.0000000262.
7. Favre E, Lion-François L, Canton M et al. Cognitive abilities of children with neurological and liver forms of Wilson disease. J Pediatr Gastroenterol Nutr 2017; 64 (3): 436–439. doi: 10.1097/MPG.0000000000001346.
8. Merle U, Schaefer M, Ferenci P et al. Clinical presentation, diagnosis and long-term outcome of Wilson’s disease: a cohort study. Gut 2007; 56 (1): 115–120.
9. Walshe JM. The acute hemolytic syndrome in Wilson’s disease – a review of 22 cases. QJM 2013; 106 (11): 1003–1008. doi: 10.1093/qjmed/ hct137.
10. Mak CM, Lam CW, Tam S. Diagnostic accuracy of serum ceruloplasmin in Wilson disease: determination of sensitivity and specificity by ROC curve analysis among ATP7B-genotyped subjects. Clin Chem 2008; 54 (8): 1356–1362. doi: 10.1373/clinchem.2008.103432.
11. Nicastro E, Ranucci G, Vajro P et al. Re-evaluation of the diagnostic criteria for Wilson disease in children with mild liver disease. Hepatology 2010; 52 (6): 1948–1956. doi: 10.1002/hep. 23910.
12. Johncilla M, Mitchell KA. Pathology of the liver in copper overload. Semin Liver Dis 2011; 31 (3): 239–244. doi: 10.1055/s-0031-1286055.
13. European Association for Study of Liver. EASL Clinical practice guidelines: Wilson’s disease. J Hepatol 2012; 56 (3): 671–685. doi: 10.1016/j.jhep.2011.11.007.
14. Weiss KH, Gotthardt DN, Klemm D et al. Zinc monotherapy is not as effective as chelating agents in treatment of Wilson disease. Gastroenterology 2011; 140 (4): 1189–1198.e1. doi: 10.1053/j.gastro.2010.12.034.
15. Ranucci G, Di Dato F, Spagnuolo MI et al. Zinc monotherapy is effective in Wilson’s disease patients with mild liver disease diagnosed in childhood: a retrospective study. Orphanet J Rare Dis 2014; 9: 41. doi: 10.1186/1750-1172-9-41.