Postižení gastrointestinálního traktu amyloidózou – kdy na ni myslet a jak diagnostikovat
Romana Ryšavá Orcid.org 1
+ Pracoviště
Souhrn
Amyloidózy jsou onemocnění odlišné etiologie, u kterých dochází k depozici abnormálně uspořádaných proteinů fibrilární ultrastruktury extracelulárně v postižených tkáních. V současné době rozeznáváme 36 různých typů amyloidóz (a spoustu jejich variant). AL amyloidóza je nejčastější formou a amyloidová depozita obsahující lehké řetězce imunoglobulinů (LC – light chain) infiltrují tkáně a způsobují jejich dysfunkci až selhání. AA a ATTR amyloidóza jsou další časté formy systémových amyloidóz. Mezi nejčastěji postižené orgány patří ledviny (v 74 %), srdce (60 %), gastrointestinální trakt (10–20 %), játra (27 %) a autonomní nervový systém (18 %). V době stanovení diagnózy má 69 % pacientů postižen více než jeden orgán. Dominantní vlastností všech typů amyloidóz je jejich barvení konžskou červení. Rozlišení jednotlivých typů amyloidóz při vyšetření biopsie ledviny či jater je založeno na přímém imunofluorescenčním vyšetření z nativních či zamrazených vzorků anebo imunohistochemicky z fixovaných (parafinových) bloků. U AL amyloidózy je dominantně pozitivní jeden z monoklonálních LC (λ nebo κ), zatímco protilátky proti druhému a ostatním fibrilárním prekurzorům jsou negativní. U AA amyloidózy se depozita barví dominantně na amyloid A, u ATTR amyloidózy na transthyretin. Gastrointestinální postižení u AL a AA amyloidózy se typicky manifestuje dysfagií, ztrátou hmotnosti, poškozením motility žaludku a střev (gastroparéza, „pseudoobstrukce“), malabsorpcí nebo krvácením. Jaterní postižení se projevuje hepatomegalií, často spojenou s portální hypertenzí. Diagnózu jaterní amyloidózy považujeme za prokázanou, pokud se v jaterní biopsii objeví depozita amyloidu pozitivně se barvící kongo červení nebo pokud má nemocný hepatomegalii větší než 15 cm či elevaci alkalické fosfatázy nad 1,5× horní hranice v dané laboratoři a současně je amyloid histologicky prokázán na jiném místě těla. Během endoskopie jsou pro amyloidózu typickými nálezy polypoidní protruze, granulární vzhled mukózy, eroze až ulcerace a submukózní hematomy. Pro optimální management nemocných s amyloidózou je nezbytná včasná diagnóza, správně stanovený typ amyloidu, efektivní léčba vč. podpůrné terapie a pečlivá monitorace nemocného během léčby.
Klíčová slova
AA amyloidóza, AL amyloidóza, hepatomegalie, malabsorpční syndrom, sérový amyloid A, volné lehké řetězceÚvod
Amyloidóza je onemocnění, pro které je charakteristické extracelulární ukládání proteinových hmot fibrilární struktury, jejichž bílkovinné prekurzory jsou chybně prostorově uspořádány a kvůli tomu dávají vzniknout proteinovým agregátům infiltrujícím tkáně. Důsledkem tohoto procesu je poškození důležitých orgánů vedoucí často k jejich selhání. Amyloidózy se běžně dělí na systémové a lokalizované formy.
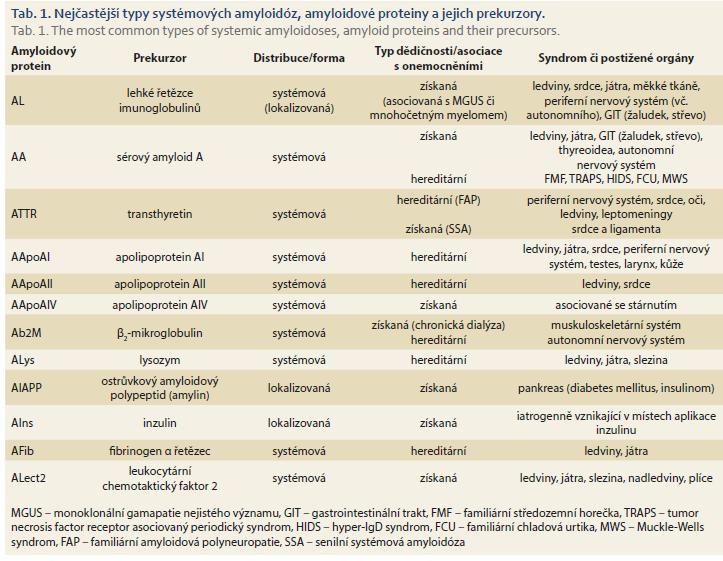
Podle příčiny zvýšené produkce prekurzoru jde o formy získané či hereditární (spojené s mutací v genu, který je zodpovědný za syntézu daného prekurzoru). Každý typ amyloidózy, ať již systémové, či lokalizované, má svůj specifický fibrilární prekurzor (amyloidogenní protein). Do dnešního dne bylo identifikováno 36 typů rozdílných fibrilárních proteinů, které mohou vést ke vzniku amyloidózy. Přehled nejčastějších systémových forem amyloidóz s možným postižením jater a gastrointestinálního traktu (GIT) je uveden v tab. 1. Označení jednotlivých typů amyloidóz se řídí určitými pravidly, která zahrnují v názvu amyloidózy zkratku A (amyloidosis) pro amyloidogenní protein a označení fibrilárního prekurzoru (např. L – immunoglobulin light chain, TTR – transthyretin). Názvosloví a dělení jednotlivých forem je pravidelně revidováno nomenklaturním výborem Mezinárodní společnosti pro amyloidózu (ISA – International Society of Amyloidosis) [1]. Kromě identifikace 36 lidských fibrilárních prekurzorů je registrováno i 10 fibrilárních zvířecích prekurzorů a 6 intracelulárních inkluzních tělísek, která jsou na rozdíl od amyloidových fibril lokalizována intracelulárně, ale vykazují řadu vlastností typických pro amyloidové fibrily (např. Lewyho tělíska, Hirano tělíska, Huntingtonova tělíska, neurofibrilární spletě).
Amyloidový fibrilární protein je definován jako protein, který je součástí amyloidových depozit ve tkáních. Amyloidová depozita se barví pozitivně konžskou červení (váže se na amyloidový protein P, který je součástí všech typů amyloidóz) a vykazují zelený dichroizmus, pokud se tato depozita obarvená konžskou červení pozorují v polarizačním mikroskopu. Chemická struktura každého proteinu musí být jasně definována přesnou sekvencí aminokyselin v proteinu a průkazem, že tvoří fibrily.
Amyloidózy jsou progresivní stavy, terapeuticky velmi špatně ovlivnitelné. Jedinou možností vyléčení je kompletní odstranění vyvolávající příčiny onemocnění, což je často prakticky nemožné (zejména u hereditárních forem).
Epidemiologie a patogeneze nejčastějších systémových forem amyloidózy
AL amyloidóza
AL amyloidóza představuje nejčastější typ systémových amyloidóz se vrůstající incidencí. Prekurzorem u tohoto typu amyloidózy jsou buď kompletní imunoglobulinové lehké řetězce (LC – light chain), nebo jejich NH2 koncové fragmenty. Amyloidogenní LC se ukládají ve tkáních, zatímco v krvi cirkulují jako volné LC (FLC – free LC). Více amyloidogenní jsou LCλ (především typ VI). Podklad nadměrné produkce monoklonálního LC tkví v existenci patologického klonu plazmatických buněk v kostní dřeni, který většinou nemá nádorový charakter, a jde tedy o monoklonální gamapatii nejasného významu (MGUS – monoclonal gammapathy of undetermined significance). AL amyloidóza komplikuje průběh mnohočetného myelomu u 5–10 % pacientů. Vyskytuje se i u řady dalších maligních hematologických onemocnění (Waldenströmova makroglobulinemie, chronická lymfatická leukemie, non-Hodgkinské lymfomy či POEMS syndrom), nicméně v 60–70 % zůstává asociována s MGUS [2]. Zatímco v letech 1950–1989 byla incidence AL amyloidózy 8,9 jedinců/mil. obyvatel, v současné době je to kolem 12 jedinců/mil. [3]. Prevalence onemocnění je ale výrazně vyšší, zejména díky delšímu přežívání pacientů s MGUS či mnohočetným myelomem. Nemocní s MGUS mají 8,8× vyšší riziko vzniku AL amyloidózy než nemocní bez MGUS [4]. Medián věku, kdy se onemocnění manifestuje, je 63 let, u osob mladších 40 let je onemocnění vzácné (< 10 % všech případů). Muži jsou o něco častěji postiženi tímto onemocněním v porovnání se ženami, a to v poměru 3: 2. Přežívání nemocných s AL amyloidózou se v poslední dekádě výrazně prodloužilo. Medián přežívání neselektované populace 868 nemocných z italské Pavie sledovaných v letech 1986–2007 byl 3,8 let [5]. U těch, kde bylo dosaženo velmi dobré odpovědi na léčbu, se medián přežívání prodloužil na 6–8 let. Pokud ale nemocní měli echokardiograficky prokázané srdeční postižení, jejich přežívání bylo jen 21 měsíců v porovnání s těmi, kteří ho neměli (82 měsíců; p < 0,001). Srdeční postižení je tak nejdůležitějším negativním prognostickým rysem pro celkové přežívání nemocných s AL amyloidózou. Ve Velké Británii je AL amyloidóza uváděna jako příčina úmrtí u 1 z 1 500 zemřelých.
AA amyloidóza
Onemocnění AA amyloidózou (dříve označované jako sekundární či reaktivní amyloidóza) vzniká jako důsledek dlouhodobého chronického zánětlivého procesu různé povahy v organizmu. Mezi nejčastější primární onemocnění, která se mohou komplikovat rozvojem AA amyloidózy, patří v našich zeměpisných šířkách revmatická onemocnění jako revmatoidní artritida, ankylozující spondylartritida, idiopatická juvenilní artritida, dále nespecifické střevní záněty (IBD – inflammatory bowel disease), chronické osteomyelitidy či bronchiektazie. Na Balkáně a v oblasti Středozemního moře to pak může být tuberkulóza či onemocnění s familiárním výskytem, jako je familiární středomořská horečka. V zemích „třetího světa“ pak mezi vyvolávající onemocnění patří i malárie a lepra, významnou roli zde hraje již zmíněná tuberkulóza. Prekurzorem cirkulujícím v séru je sérový amyloid A (SAA). Chronický zánětlivý proces vede prostřednictvím stimulace monocyto-makrofágového systému ke zvýšení sekrece interleukinu 1 a 6 a tumor nekrotizujícího faktoru α. Játra na tento podnět reagují zvýšením sekrece proteinů akutní fáze, vč. SAA. Hladiny SAA v krvi se během zánětu rychle zvyšují (až 1 000× oproti normálu), velmi dobře korelují s ostatními zánětlivými parametry, a to jak se sedimentací erytrocytů, tak s hladinou C-reaktivního proteinu (CRP). Přesné údaje o výskytu AA amyloidózy v neselektované populaci nemocných jsou velmi obtížně zjistitelné. Ve starších publikacích se výskyt odhadoval na 4 pacienty/100 000 obyvatel [6]. V současné době se výskyt AA amyloidózy výrazně snížil v důsledku zavedení biologické léčby do terapie jak revmatologických onemocnění, tak např. u pacientů s IBD. Přibylo naopak případů, kde AA amyloidóza vzniká jako důsledek chronických infekcí (dekubity, osteomyleitidy) či jako komplikace hidradenitidy. Bohužel stoupá i počet pacientů, u kterých nejsme schopni ani za pomoci nejnovějších diagnostických metod určit základní, vyvolávající onemocnění (až v 18 % případů) [7]. Medián vzniku AA amyloidózy od prvních příznaků základního onemocnění je velmi variabilní a může to být několik měsíců, ale i 20 let. Přibližně 10 % nemocných s revmatoidní artritidou umírá na komplikace, které souvisejí s přítomností amyloidózy a/nebo na infekční komplikace spojené s léčbou [8]. Medián přežití nemocných s AA amyloidózou se v současné době pohybuje kolem 133 měsíců a významně závisí na koncentraci SAA [9]. Pokud se podaří léčbou udržet koncentraci SAA < 4 mg/l, dramaticky se tím prodlouží doba přežití v porovnání s jedinci, u kterých jsou koncentrace SAA > 155 mg/l (až 18×). Podobná data přinesla i starší studie ze stejného pracoviště, kde koncentrace SAA v séru < 10 mg/l byla spojena s regresí depozit amyloidu ve tkáních a 10leté přežívání bylo 90 %, zatímco dlouhodobá koncentrace SAA > 50 mg/l vedla k nárůstu amyloidových depozit a 10leté přežívání bylo jen 40 % (rozdíl v přežívání mezi skupinami byl významně signifikantní; p = 0,0009) [10].
ATTR amyloidóza
Nejčastější formou hereditárních amyloidóz je ATTR amyloidóza. Odhadovaná prevalence onemocnění celosvětově je 1/100 000 obyvatel. Výskyt velmi kolísá v závislosti na regionu, kdy je několik endemických oblastí s velmi vysokou prevalencí onemocnění až 1/1 000 obyvatel (Portugalsko, Švédsko, Japonsko) [11]. Tato amyloidóza je vyvolána ukládáním fibrilárních depozit TTR (protein fungující jako transportér hormonů thyreoidey a retinolu) ve tkáních a může se vyskytovat jako získaná forma (SSA; způsobuje ji divoký typ TTR) nebo jako hereditární forma, kde mutace v genu pro TTR vedou ke vzniku jeho různých variant, které jsou amyloidogenní. Klinickým korelátem tohoto stavu je onemocnění, které je běžně označováno jako familiární amyloidová polyneuropatie (FAP), podle současné nomenklatury ATTR amyloidóza.
AApoAI amyloidóza
Pravděpodobně druhou nejčastější hereditární amyloidózou je AApoAI amyloidóza [12]. Fibrilárním prekurzorem je apolipoprotein AI, který ale nebývá kompletní a z původního řetězce o 243 aminokyselinách se na vzniku amyloidové fibrily podílejí fragmenty s 93 aminokyselinami. Primární struktura ApoAI je uspořádána do α-helixu, a tudíž vytvoření β-struktury skládaného listu (typické pro amyloidózu) vyžaduje poměrně složitou terciární přestavbu v prostoru. Fenotyp onemocnění je závislý na lokalizaci mutace. Je-li mutace uložena na aminoterminální části proteinu (postihuje aminokyseliny 1–75), projevuje se onemocnění dominantním postižením ledvin a jater. Proteinurie u tohoto typu amyloidu nebývá velká, jelikož depozita postihují zejména dřeň ledviny a ne glomeruly. Postižení jater je často vedlejším nálezem při provádění jaterní biopsie z jiných důvodů nebo např. při cholecystektomii. Z dalších klinických projevů stojí za zmínku periferní neuropatie či kardiální postižení.
AFib amyloidóza
AFib amyloidóza, jejímž fibrilárním prekurzorem je Aα řetězec fibrinogenu, se typicky projevuje postižením glomerulů s masivní proteinurií [13]. Jde o poměrně rychle progredující onemocnění, často doprovázené hypertenzí. Játra bývají nezřídka také postižena. V amyloidových depozitech se ukládá pouze fragment proteinu, a to jeho C-terminální část, což je důvodem, že imunohistochemický průkaz tohoto typu amyloidózy může být často svízelný.
Klinické projevy a subjektivní příznaky amyloidózy
Vlastní projevy nemoci jsou velmi pestré a individuálně značně rozdílné. Pravidelnou součástí klinického obrazu jsou jak celkové nespecifické projevy jako slabost, malátnost, únavnost, snížení chuti k jídlu a úbytek hmotnosti, tak pestré projevy vyplývající z postižení jednotlivých orgánů a tkání.
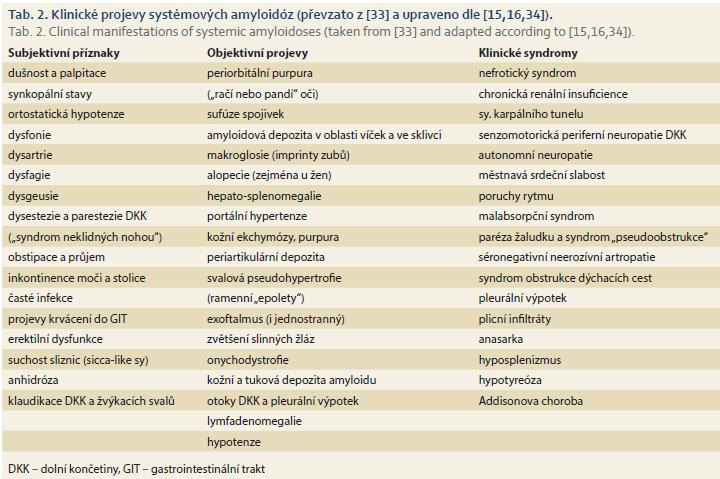
Nejčastější projevy systémových amyloidóz jsou uvedeny v tab. 2.
Systémové amyloidózy většinou postihují více orgánů současně, ale míra jejich poškození může být různá a závisí jak na typu proteinového prekurzoru (např. AL amyloidóza λ nejčastěji infiltruje ledviny a způsobuje nefrotický syndrom, zatímco přítomnost κ řetězců bývá spojena s extrarenálním, zejména GIT postižením), tak na jeho afinitě k určitému orgánu a na lokálních poměrech v dané tkáni. Postižení jednoho orgánu v době stanovování diagnózy se u AL amyloidózy vyskytuje ve 25 %, dvou v 36 %, třech a více orgánů u 39 % nemocných [14]. Postižení ledvin se vyskytuje v 72 %, kardiomyopatie v 63 %, postižení jater v 27 %, periferní neuropatie v 19 %, postižení autonomního nervstva v 16 %, postižení měkkých tkání v 12 % a amyloidová kožní purpura v 11 % [14]. U nemocných s AA amyloidózou bývá postižení GIT častější a vyskytuje se až u 60 % nemocných.
Gastrointestinální a hepatální příznaky amyloidózy
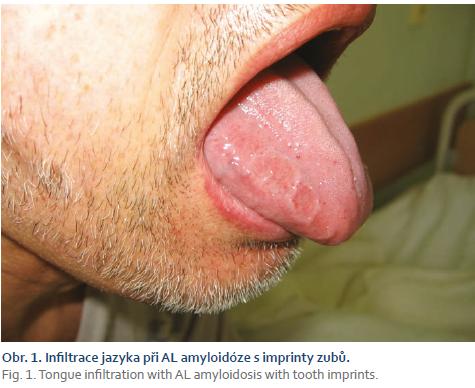
Systémové amyloidózy kromě ledvin a srdce velmi často postihují i orgány GIT a játra. Infiltrace jazyka amyloidovými depozity se manifestuje jako makroglosie a vyskytuje se asi u 10 % nemocných (obr. 1). Je-li extrémní, může vést až k problémům s dýcháním v důsledku obstrukce dýchacích cest, jelikož infiltruje zejména kořen jazyka. Infiltrace sliznice žaludku může způsobit gastroparézu s poruchou jeho vyprazdňování a opakovaným zvracením, pocity předčasné sytosti a plnosti žaludku. Při postižení sliznice tenkého střeva amyloidem (zejména u AA amyloidózy) trpí nemocní malabsorpčním syndromem a často postprandiálními průjmy; u AL amyloidózy se naopak objevuje obstipace (pseudoobstrukce), která bývá způsobena depozity amyloidu v muscularis mucosae a postižením cévní vaskulatury střeva amyloidem. V rámci amyloidózy GIT byla popsána i perforace střeva, střevní infarzace a výrazné krvácení z distálních oblastí zažívací trubice jako důsledek zvýšené fragility cév [15]. Depozita v játrech způsobují hepatomegalii, která může v pokročilejších stadiích onemocnění vést k rozvoji portální hypertenze (až u 1/4 nemocných). Játra jsou často dobře hmatná a zvětšená v rozsahu několika centimetrů pod oblouk žeberní. Nemocní si nezřídka stěžují na tlakové bolesti v pravém hypochondriu způsobené tlakem zvětšených jater na jejich pouzdro. Rovněž slezina bývá časně infiltrována amyloidem, což vede k jejímu zvětšení a tlakovým bolestem v levém hypochondriu. Hyposplenizmus může být jedním z důvodů, který přispívá k častým infekcím u pacientů [16].
Diagnostika amyloidózy
Amyloidóza je onemocnění, kde diagnostika musí být založena na histologickém průkazu depozit amyloidu ve tkáních. V současné době není k dispozici metoda, která by nahradila histologický průkaz amyloidu. Ani ten nemusí být vždy jednoduchý, protože variabilita ukládání amyloidových depozit ve tkáních a nekonzistentnost barvení depozit pomocí imunohistochemických metod mohou často způsobit jejich přehlédnutí, zejména jde-li o časná stadia onemocnění.
Biomarkery u amyloidózy
U řady amyloidóz, zejména hereditárních, bývají koncentrace měřeného prekurzoru v séru normální a amyloidová depozita tvoří jejich fragmenty přímo ve tkáních. U získaných amyloidóz jako AA či AL ale můžeme detekovat zvýšené koncentrace prekurzorů v séru, čehož se využívá nejen k detekci typu amyloidu, ale zejména k monitoraci aktivity onemocnění.
U AA amyloidózy je nutné stanovit koncentraci SAA v séru. Hodnoty pod 10 mg/l jsou obecně považovány za normální, resp. nerizikové pro rozvoj AA amyloidózy. O vlivu jeho koncentrací na prognózu nemocných již bylo pojednáno výše.
Průkaz přítomnosti monoklonálního proteinu (MP) tvořeného LC v séru u AL amyloidózy se opírá o některé klíčové testy. Elektroforéza sérových bílkovin je ale hrubě orientační metodou pro průkaz MP a až ve 40 % případů nemusí detekovat jeho přítomnost. U všech nemocných by proto měla být provedena imunofixace (IFE – immunofixation electrophoresis) séra, která je schopna detekovat přítomnost MP s vysokou senzitivitou (95 %) a dále identifikuje typ MP. Nevýhodou metody je to, že poskytuje jen kvalitativní data a nezískáme informaci o koncentraci MP v séru [17]. Proto tyto testy doplňujeme o stanovení koncentrace volných LC v séru (sFLC), což je vyšetření schopné odhalit přítomnost MP v malých koncentracích (kolem 10 mg/l). V současné době jsou dostupné dva testy, které detekují sFLC buď pomocí polyklonálních (FreeliteTM, Birmingham, Velká Británie), nebo monoklonálních protilátek proti LC (N-latex, Siemens, Marburg, Německo) [18,19]. Testy měří koncentrace obou izotypů FLC (κ i λ) a současně počítají i jejich poměr (κ/λ ratio). Tento poměr se významně zvyšuje nad normu či pod normu (0,26–1,65 u metody Freelite) v případě klonální produkce některého z řetězců. Kombinace IFE a sFLC má vysokou senzitivitu pro průkaz MP (až 98 %). Určitá opatrnost při interpretaci výsledků je nutná v případě, že má nemocný sníženou glomerulární filtraci (GF), jelikož FLC se u těchto nemocných v organizmu retinují a zvyšuje se nejen jejich absolutní koncentrace, ale i poměr [20].
Postižení ledvin prokazujeme pomocí stanovení proteinurie/24 hod nebo vyšetřením poměru protein/kreatinin v jednorázovém vzorku moči. Typickým nálezem je nefrotická proteinurie (> 3,5 g/den); řada nemocných má plně vyjádřený nefrotický syndrom, proto je nutné vyšetřit i hladinu sérového albuminu a cholesterolu. Standardní součástí vyšetření moči je i močová IFE. Průkaz FLC v moči je vhodný zejména v situaci, kdy sFLC jsou v normě a sérová IFE je negativní. Snížení GF se objevuje většinou až v pozdějších stadiích poškození ledvin a dnes se preferuje používání výpočtových metod k odhadu GF (eGF – estimated GF), jako jsou CKD-EPI či MDRD.
Postižení jater se projevuje zejména vzestupem hodnot alkalické fosfatázy (ALP – alkaline phosphatase) a gamaglutamyltransferázy (GMT). Bilirubin bývá zvýšený až při pokročilejších stadiích jaterního poškození a často se objevuje spolu se známkami portální hypertenze.
Pro srdeční postižení je typický vzestup N-terminal pro-brain natriuretic peptidu (NT-proBNP) nebo BNP a srdečních troponinů (tab. 3). Nověji popisovaným markerem srdečního a ledvinného postižení je růstový diferenciační faktor 15, jehož vysoké hodnoty jsou jednoznačně spojovány s vyšším rizikem úmrtí a celkově horším přežíváním nemocných [21]. Vhodné je stanovit i hormony štítné žlázy, jelikož jak AA, tak zejména AL amyloidóza mohou infiltrovat tyreoideu a způsobit hypotyreózu.
Zobrazovací metody
Využití zobrazovacích metod u amyloidózy sleduje dva hlavní aspekty, tj. morfologické a funkční posouzení orgánového postižení amyloidem (zejména myokardu). K detekci amyloidových depozit v organizmu lze využít i scintigrafické vyšetření. Radiografické vyšetření (RTG) v diagnostickém algoritmu samotné amyloidózy nemá větší diagnostický přínos a dominantně je využíváno k vyloučení osteolytického postižení skeletu při asociaci systémové AL amyloidózy s mnohočetným myelomem. Pomocí RTG hrudníku lze prokázat rozšíření srdečního stínu s městnáním v malém oběhu, pleurální výpotek a vzácně postižení plic při amyloidóze. CT vyšetření má u systémové amyloidózy limitovaný význam zejména z důvodu nízké specificity a hlavní využití představují ložiskové formy onemocnění zejména v oblasti orbity, nazofaryngu či laryngu. Dominantně je však CT vyšetření využíváno k detekci amyloidózy v oblasti tracheobronchiálního stromu (fokální nebo difuzní submukózní depozita zobrazující se jako noduly, plaky či cirkulární ztluštění stěny) a k posouzení postižení plicního parenchymu (nodulární parenchymatózní amyloidóza nebo difuzní alveolární septální amyloidóza). CT vyšetření břicha je přínosné v diagnostice a diferenciální diagnostice jaterního postižení, přičemž diagnostická kritéria vyžadují hepatomegalii > 15 cm. Ultrazvukové vyšetření (UZ) se využívá zejména v diagnostice srdečního poškození u AL a ATTR amyloidózy při echokardiografii (ECHO). ECHO je v současnosti stěžejní metodou pro diagnostiku a sledování srdečního postižení při amyloidóze. Kritériem postižení je koncentrická hypertrofie myokardu levé komory s diastolickou šíří interventrikulárního septa > 12 mm při absenci hypertenze nebo jiné příčiny hypertrofie myokardu [22]. Dalšími ECHO nálezy jsou diastolická dysfunkce s restriktivním plněním levé komory, snížená ejekční frakce, ztráta longitudinální kontrakční schopnosti levé komory, dilatace levé síně a přítomnost chlopenních vad. Typickým nálezem je sonografický obraz jemně granulovaného myokardu („granular sparkling“) [23]. Přínosem může být u nemocných s jaterním postižením elastografie, kdy masivní depozita amyloidu bývají spojena s nálezem vysoké tuhosti jater (fibrózy). Magnetická rezonance (MR) je stejně jako v případě UZ vyšetření využívána zejména v hodnocení srdečního postižení. Fenomén pozdního sycení („delayed enhancement“) ve své difuzní subendokardiální či méně časté transmurální formě zobrazení je nález charakteristický pro postižení myokardu amyloidem [24]. MR je užitečná i v posuzování ložiskové amyloidózy v oblasti laryngu, nazofaryngu, míchy či urogenitálního traktu. Radioscintigrafie s využitím značeného 123I-SAP (serum amyloid P component) je neinvazivní, kvantitativní a velmi citlivou metodou (u AL a AA amyloidózy 90% senzitivita) pro celotělovou detekci orgánové distribuce amyloidu s dobrou vizualizací depozit v ledvinách, játrech, kostech, slezině a nadledvinkách, a tím i vhodnou technikou k monitorování výsledků léčby. 123I-SAP scintigrafie umožňuje vizualizaci amyloidových depozit i v orgánech bez známek klinického postižení a v případě nemožnosti biopsie [25,26]. Celosvětově nízká dostupnost 123I-SAP je podmíněna potenciálním rizikem přenosu infekce, neboť SAP se připravuje z krve dárců. V ČR není tato metoda zavedena. V současnosti je na některých pracovištích užíváno 99mTc-3,3-diphosphono-1-2-propanodicarboxylic acid (DPD) scintigrafie. 99mTc-DPD je selektivně vychytáván v myokardu s depozity TTR amyloidu, což je výhodné nejen pro diagnostiku a posuzování postižení srdce u hereditární a senilní ATTR amyloidózy, ale i pro diferenciální diagnostiku vůči AL, u které akumulace radiofarmaka chybí či je velmi nízká [27]. Tato metoda je dostupná i v ČR. Endoskopická vyšetření (gastroskopie, koloskopie, rektoskopie) nemívají specifický obraz amyloidózy, nicméně mohou upozornit na poruchy spojené s postižením jater, např. na přítomnost portální hypertenze (portální gastropatie, jícnové varixy). Depozita amyloidu přímo ve sliznici či v podslizničních oblastech GIT mohou vypadat jako mnohočetné polypoidní protruze, slizniční eroze, ulcerace a submukózní hematomy [28]. Sliznice bývá často velmi fragilní, granulárního vzhledu či s nálezem difuzních petechií a náchylná ke krvácení. V oblasti tlustého střeva se mohou depozita amyloidu manifestovat jako kolitida s ulceracemi.
Histologický průkaz amyloidu
Suverénní metodou průkazu amyloidózy je nález kongo pozitivních hmot při vyšetření bioptického vzorku. Při barvení konžskou červení se amyloidová depozita jeví jako oranžovo-červené okrsky amorfní hmoty bez hypercelularity. Orientační rozdělení vzorků na AA a AL amyloid umožňuje předchozí oxidace preparátů manganistanem draselným. AA amyloid po vystavení účinku manganistanu a následném obarvení konžskou červení ztrácí charakteristický dichroizmus (zelený dvojlom) při pozorování v polarizačním mikroskopu. AL amyloid si tuto vlastnost zachovává. Správné výsledky barvení jsou do značné míry závislé na síle histologických řezů a zkušenosti vyšetřujícího personálu, a proto lze dnes tento způsob identifikace základních typů amyloidu považovat za hrubě orientační. Dříve se k průkazu amyloidových depozit používaly i další metody jako reakce s krystalovou (metylovou) violetí, kdy se depozita barvila fialovo-červeně, či reakce s tioflavinem S a T, kde depozita vykazovala zeleno-žlutou fluorescenci. Tato poslední metoda je sice poměrně citlivá, ale není specifická pouze pro amyloidózu. I proto se obě tyto metody dnes již považují za zastaralé.
V současné době se používají pro průkaz jednotlivých typů amyloidu specifičtější metody, a to zejména imunofluorescenční vyšetření nativních či zmrazených vzorků. Zde se standardně využívají protilátky proti různým komponentám (např. imunoglobulinům či složkám komplementu), ale také proti AA amyloidu a LC κ a λ. Zejména pro průkaz AL amyloidózy jde o klíčové vyšetření [29]. Imunohistochemické vyšetření z již fixovaných (parafinových) vzorků lze použít pro průkaz AA amyloidózy, AB2M či ATTR amyloidózy, detekce LC a průkaz AL amyloidózy jsou ale touto metodou méně spolehlivé (z důvodu snížení schopnosti LC navázat protilátku po zalití do parafinu a dále také z důvodu značné přirozené variability N-terminálních úseků LC). V případě nejasnosti o typu amyloidu (pozitivní barvení konžskou červení a negativní či neurčitá imunofluorescence či imunohistochemie s běžně používanými protilátkami) je nově možné detekovat amyloidová depozita pomocí hmotnostní spektrometrie. Amyloidová depozita (většinou 3–4 ze vzorku) se označí na histologickém preparátu a pomocí laserové mikrodisekce se vyříznou z bloku. Následně se pomocí proteomické analýzy určí sekvence aminokyselin obsažených v proteinu a ta se porovná s databází známých proteinů pomocí speciálního softwaru. Tím se identifikuje protein, který tvoří amyloidová depozita. Správnost výsledku by se měla opírat i o fakt, že depozita jakéhokoli amyloidu obsahují amyloidový protein P a apolipoproteiny E a J, které by měly být ve vzorku detekovány společně s fibrilárním prekurzorem a ve stejné lokalitě [30]. Nález amyloidových fibril v elektronové mikroskopii je potvrzením přítomnosti amyloidózy; fibrily mají náhodné uspořádaní, jsou nevětvené, 8–10 nm široké a 1 μm dlouhé, a následně tvoří jakousi síť.
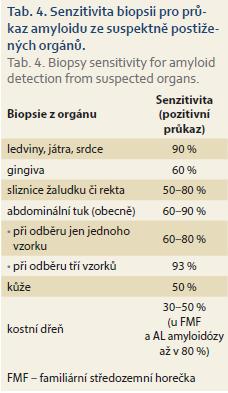
K histologickému průkazu přítomnosti amyloidózy lze použít vzorek tkáně, který získáme při nekropsii, během operačních výkonů či z biopsie suspektně postiženého orgánu. Mezi tkáně, které se používají k průkazu amyloidových depozit při necílených biopsiích, patří gingiva, žaludeční sliznice, rektální sliznice, abdominální tuk (vzhledem k nižší frekvenci postižení těchto tkání amyloidovými depozity může být výsledek falešně negativní, proto se standardně doporučuje odebrat 3 vzorky k vyšší záchytnosti vyšetření) [31], dále biopsie ledvin, jater či biopsie endomyokardiální – frekvence postižení těchto orgánů je podstatně vyšší, tudíž i záchyt amyloidových depozit je pravděpodobnější (tab. 4). U nemocných s neuropatií je možné provést biopsii surálního nervu (zejména při podezření na FAP). Průkaz amyloidových depozit v nervových strukturách vždy signalizuje již pokročilé stadium onemocnění. Počáteční stadia onemocnění se často detekují zejména v cévní stěně postižených orgánů, a proto vykazují orgány s větší sítí cév (ledviny, játra) vyšší senzitivitu pro průkaz amyloidu. Na tomto místě je tedy nutné zdůraznit, že prokážeme-li histologicky depozita amyloidu (u systémových amyloidóz) v některém orgánu, je velmi pravděpodobné, že amyloid bude i v těch dalších. Současně je třeba mít na paměti, že v případě negativní histologie nemůžeme přítomnost amyloidózy vyloučit, protože při biopsii odebereme jen malý okrsek tkáně a žádná z bioptických metod nemá 100% senzitivitu [32]. Proto je nutné u jedinců, kde máme naléhavé podezření na amyloidózu a kteří jsou současně ve vysokém riziku vzniku onemocnění, biopsii v časovém odstupu opakovat či zvolit pro průkaz biopsii jiného suspektně postiženého orgánu, kde může být depozit více.
Jak lze tedy definovat nezbytné minimum vyšetření, pomocí nichž bychom měli dospět k diagnóze amyloidózy?
- sérum: elektroforéza bílkovin IFE, koncentrace FLC a poměr κ/λ; SAA, CRP; troponin T a I, NT-proBNP; ALP, GMT;
- moč: proteinurie/24 hod (či poměr protein/kreatinin ze vzorku moči), IFE moči, fakultativně FLC v moči;
- EKG, EKG-Holter, ECHO, (MR srdce);
- bioptická verifikace typu amyloidu z dominantně postiženého orgánu nebo necílená biopsie z abdominálního tuku nebo rekta/gingivy/jazyka;
- trepanobiopsie; RTG či low-dose CT skeletu v případě potvrzení AL amyloidózy;
- elektromyografie dolních končetin;
- při průkazu jiného typy amyloidu než AL či AA hmotnostní spektrometrie z amyloidových depozit s určením typu amyloidózy a dle výsledku genetické vyšetření na průkaz mutace.
Závěr
I přes pokrok v diagnostice a léčbě patří všechny systémové formy amyloidóz k závažným stavům, které je třeba rychle a dobře diagnostikovat a po identifikaci typu amyloidu zahájit cílenou léčbu. Stále se bohužel setkáváme s pacienty, kdy je diagnóza stanovena pozdě a plnohodnotnou léčbu si již nemůžeme dovolit. Na onemocnění je potřeba myslet a při nejasných diagnostických závěrech žádat patology o provedení barvení na kongo červeň (ne vždy se standardně provádí). Gastroenterologové se zejména mohou setkat s nemocnými, kde se amyloidóza projevuje jako hepatopatie nejasného původu s elevací obstrukčních jaterních testů bez zvýšení bilirubinu, hepatomegalií a nakonec s příznaky portální hypertenze (již pokročilá stadia onemocnění).
Doručeno/Submitted: 27. 2. 2019
Přijato/Accepted: 9. 4. 2019
prof. MUDr. Romana Ryšavá, CSc.
Klinika nefrologie
1. LF UK a VFN v Praze
U Nemocnice 2
128 08 Praha 2
romana.rysava@vfn.cz

Pro přístup k článku se, prosím, registrujte.
Výhody pro předplatitele
Výhody pro přihlášené
Literatura
1. Benson MB, Buxbaum JN, Eisenberg DS at al. Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 2018; 25 (4): 215–219. doi: 10.1080/13506129.2018.1549825.
2. Rysava R. AL amyloidosis: advences in diagnostics and treatment. Nephrol Dial Transplant 2018. In press. doi: 10.1093/ndt/gfy291.
3. Duhamel S, Mohty D, Magne J et al. Incidence and prevalence of light chain amyloidosis: a population-based study. Blood 2017; 130: 5577.
4. Kyle RA, Larson DR, Therneau TM et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. N Engl J Med 2018; 378 (3): 241–249. doi: 10.1056/NEJMoa1709974.
5. Merlini G, Palladini G. Amyloidosis: is a cure possible? Ann Oncol 2008; 19 (Suppl 4): iv63–66. doi: 10.1093/annonc/mdn200.
6. Hazenberg BP, van Rijswijk MH. Clinical and therapeutic aspects of AA amyloidosis. Baillieres Clin Rheumatol 1994; 8 (3): 661–668.
7. Lachmann HJ, Gillmore JD, Wechalekar AD et al. A single centre 20 year case series of AA amyloidosis – changing epidemiology. XIIIth International Symposisum on Amyloidosis, Groningen 2012; OP 51.
8. Hazes, JM, Cats A. Rheumatoid arthritis. In: Klippel JH, Dieppe PA (eds). Rheumatology. Mosby-Year Book Europe Limited 1994.
9. Lachmann HJ, Goodman HJ, Gilbertson JA et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med 2007; 356 (23): 2361–2371.
10. Gillmore JD, Lovat LB, Persey MR et al. Amyloid load and clinical outcome in AA amyloidosis in relation to circulating concentration of serum amyloid A protein. Lancet 2001; 358 (9375): 24–29.
11. Sousa A, Coelho T, Barros J et al. Genetic epidemiology of familial amyloidotic polyneuropathy (FAP) -type I in Póvoa do Varzim and Vila do Conde (north of Portugal). Am J Med Genet 1995; 60 (6): 512–521.
12. Nichols WC, Dwulet FE, Liepnieks J et al. Variant apolipoprotein AI as a major constituent of a human hereditary amyloid. Biochem Biopsys Res Commun 1988; 156 (2): 762–768.
13. Benson MD, Liepnieks J, Uemichi T et al. Hereditary renal amyloidosis associated with a mutant fibrinogen alpha-chain. Nat Genet 1993; 3 (3): 252–255.
14. Merlini G. AL amyloidosis: diagnosis and prognosis. Haematologica 2007; 92 (6) (Suppl 2): 58–59.
15. Guidelines Working Group of UK Myeloma Forum, British Commitee for Standards in Haematology, British Society for Haematology. Guidelines on the diagnosis and management of AL amyloidosis. Br J Haematol 2004; 125 (6): 681–700.
16. Ryšavá R. Systémové amyloidózy a jejich léčba. Praha: Maxdorf Jessenius 2013.
17. Yadav P, Leung N, Sanders PW et al. The use of immunoglobulin light chain assays in the diagnosis of paraprotein-related kidney disease. Kidney Int 2015; 87 (4), 692–697. doi: 10.1038/ki.2014.333.
18. Bradwell AR, Carr-Smith HD, Mead GP et al. Highly sensitive, automated immunoassay for immunoglobulin free light chains in serum and urine. Clin Chem 2001; 47 (4): 673–680.
19. te Velthuis H, Knop I, Stam P et al. N Latex FLC – new monoclonal high-performance assays for the determination of free light chain kappa and lambda. Clin Chem Lab Med 2011; 49 (8): 1323–1332. doi: 10.1515/CCLM.2011.624.
20. Hutchison CA, Basnayake K, Cockwell P. Serum free light chain assessment in monoclonal gammopathy and kidney disease. Nat Rev Nephrol 2009; 5 (11): 621–628. doi: 10.1038/nrneph.2009.151.
21. Kastritis E, Papassotiriou I, Merlini G et al. Growth differentiation factor-15 is a new biomarker for survival and renal outcomes in light chain amyloidosis. Blood 2018; 131 (14): 1568–1575. doi: 10.1182/blood-2017-12-819904.
22. Gertz MA, Comenzo R, Falk RH et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International symposium on amyloid and amyloidosis, Tours, France, 18–22 April 2004. Am J Hematol 2005; 79 (4): 319–328.
23. Fikrle M, Paleček T, Kuchyňka P et al. Cardiac amyloidosis: a comprehensive review. Cor et Vasa 2013; 55 (1): e60–75.
24. Maceira AM, Joshi J, Prasad SK et al. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation 2005; 111 (2): 186–193. doi: 10.1161/01.CIR.0000152819.97857.9D.
25. Hawkins PN. Serum amyloid P component scintigraphy for diagnosis and monitoring amyloidosis. Curr Opin Nephrol Hypertens 2002; 11 (6): 649–655.
26. Palladini G, Perlini S, Merlini G. Imaging of systemic amyloidosis. In: Gertz MA, Rajkumar SV (eds). Amyloidosis: diagnosis and treatment. Springer Science and Business Media, LLC 2010; 15–32. doi: 10.1007/978-1-60761-631-3_2.
27. Rapezzi C, Quarta CC, Guidalotti PL et al. Role of (99m) Tc-DPD scintigraphy in diagnosis and prognosis of hereditary transthyretin-related cardiac amyloidosis. JACC Cardiovasc Imaging 2011; 4 (6): 659–670. doi: 10.1016/j.jcmg.2011.03.016.
28. Koop AH, Mousa OY, Wang MH. Clinical end endoscopic manifestation of gastrointestinal amyloidosis: a case series. Clujul Medical 2018; 91 (4): 469–473. doi: 10.15386/cjmed-951.
29. Said SM, Sethi S, Valeri AM et al. Renal amyloidosis: origin and clinicopathologic correlation of 474 recenet cases. Clin J Am Soc Nephrol 2013; 8 (9): 1515–1523. doi: 10.2215/CJN.10491012.
30. Sethi S, Theis JD, Leung N et al. Mass spectrometry-based proteomic diagnosis of renal immunoglobulin heavy chain amyloidosis. Clin J Am Soc Nephrol 2010; 5 (12): 2180–2187. doi: 10.2215/CJN.02890310.
31. van Gameren II, Hazenberg BP, Bijzet J et al. Diagnostic accuracy of subcutaneous abdominal fat tissue aspiration for detecting systemic amyloidosis, and its utility in clinical practice. Arthritis Rheum 2006; 54 (6): 2015–2021.
32. Picken MM. Amyloidosis – where are we now and where are we heading? Arch Pathol Lab Med 2010; 134 (4): 545–551. doi: 10.1043/1543-2165-134.4.545.
33. Pika T et al. Diagnostika a léčba systémové AL amyloidózy. Transfuze Hematologie dnes 2019. In press.
34. Gertz MA, Hayman SR. Immunoglobulin light chain amyloidosis. In: Rajkumar SV, Kyle RA. Treatment of multiple myeloma and related disorders. Cambridge: Cambridge University Press 2009: 112–128.
35. Gertz MA, Merlini G. Definition of organ involvement and response to treatment in AL amyloidosis: an updated consensus opinion. Amyloid 2010; 17: 48–49.