Nemoc ze střádání esterů cholesterolu - efekt dlouhodobé léčby lovastatinem
Vratislav Šmolka1
+ Pracoviště
Souhrn
Deficit lyzozomální kyselé lipázy se manifestuje ve dvou prognosticky odlišných klinických variantách: maligní infantilní varianta (Wolmanova nemoc) a relativně benigní forma, která je známa jako nemoc ze střádání esterů cholesterolu (CESD). V kazuistice prezentujeme dívku s hepatomegalií, poruchou růstu, neprospíváním a hyperlipidémií. Podezření na CESD bylo potvrzeno histologickým vyšetřením jater a výrazným snížením aktivity enzymu kyselé lipázy v leukocytech. U pacientky byla ve dvou letech věku zahájena léčba lovastatinem, inhibitorem 3-hydroxy-3-methylglutaryl koenzym A reduktázy. V průběhu pěti let trvající léčby klesla hladina cholesterolu, triglyceridů a LDL cholesterolu. Váha a výška se upravily a odpovídají průměrným hodnotám pro daný věk. Hladina HDL cholesterolu se nezměnila. Sérové hladiny transamináz v průběhu léčby kolísaly a velikost jater se při ultrasonografickém vyšetření nezměnila. Léčba lovastatinem probíhá bez klinických komplikací. Prognóza jaterního postižení zůstává však do budoucna nejasná.
CESD patří do diferenciální diagnostiky hepatomegalie od kojeneckého věku až po dospělé pacienty. Léčba lovastatinem je dobře tolerovaná, snižuje hladiny tuků, ale vliv statinů na progresi jaterního postižení zůstává nejasný.
Klíčová slova: nemoc ze střádání esterů cholesterolu - lovastatin.
ÚVOD
Nemoc ze střádání esterů cholesterolu (cholesteryl ester storage disease - CESD) je vzácné dědičné onemocnění, jehož příčinou je deficit lyzozomální kyselé lipázy (LKL - acid cholesterol hydrolyse, EC 3.1.1.13). Enzym je přítomen v lyzozymech jaderných buněk a katalyzuje hydrolýzu esterů cholesterolu a triglyceridů. Deficit LKL vede ke střádání tuků v tkáních s vysokou receptorově zprostředkovanou endocytózou lipoproteinů s nízkou denzitou (LDL). Klinicky se nejčastěji projeví hepatomegalií. Pacienti mají hypercholesterolémii, někteří hypertriglycerolémii, vysokou hladinu LDL cholesterolu, nízkou hladinu lipoproteinů o vysoké denzitě (HDL - high density lipoprotein) a jsou ohroženi předčasným vznikem aterosklerózy. Kromě této relativně benigní formy deficitu LKL je známa maligní infantilní varianta (Wolmanova nemoc). Onemocnění se klinicky manifestuje v prvním až druhém měsíci života zvracením, průjmy, neprospíváním, hepatosplenomegalií, oboustrannou kalcifikací nadledvin. Většina pacientů umírá do jednoho roku života. Určujícím faktorem rozsahu postižení je pravděpodobně výše reziduální aktivity enzymu(1). Obě nemoci jsou dědičné autosomálně recesivně. Strukturální gen pro LKL je lokalizován v oblasti dlouhých ramen chromosomu 10 v pruhu q24-q25. Pro obě nemoci jsou známy prevalentní mutace(2). Neexistuje žádná kauzální léčba obou nemocí. Při podávání inhibitoru 3-hydroxy-3-methylglutaryl koenzym A (HMG-CoA) reduktázy (statinů) bylo popsáno snížení hladiny cholesterolu a triglyceridů u CESD(3,4).
KAZUISTIKA
Pacientka je z prvního fyziologického těhotenství nepříbuzných rodičů. Porod byl v termínu, porodní váha 3600 g a porodní délka 51 cm. Adaptace proběhla bez komplikací. Dítě bylo plně kojeno 6 měsíců. V 1 roce byla váha dítěte 7800 g (3 percentil), délka 73 cm (25 percentil). V 18. měsíci věku byla pacientka hospitalizována na infekčním oddělení pro salmonelovou gastroenteritidu. Pro neprospívání byla ve 21. měsíci věku vyšetřena v dětské gastroenterologické poradně.
Při vyšetření byla zjištěna hepatomegalie, vyšší hladiny transamináz a hypercholesterolémie. Nebyly prokázány protilátky proti gliadinu a endomyziální protilátky. Opakovaná kultivační vyšetření stolic neprokázala salmonelovou infekci. Při doporučeném přijetí na Dětskou kliniku v Olomouci ve 23. měsíci věku byla váha dítěte 10,5 kg (10 percentil) a výška 83 cm (10-25 percentil). Pacientka byla dystrofická s minimem podkožního tuku, játra byla 4 cm pod žeberním obloukem. Ostatní klinický nález byl bez patologického nálezu. Z biochemického vyšetření byla hladina ALT 1,69 μkat/1, AST 1,5 μkat/1, cholesterolu 7,57 mmol/1, triglyceridů 6,1 mmol/1, HDL cholesterolu 0,67 mmol/1 a LDL cholesterolu 6,0 mmol/1. Všechny ostatní výsledky biochemických vyšetřeních (minerály, celková bílkovina, albumin, praealbumin, transferin, cholinesteráza, bilirubin, gama-glutamyltransferáza, laktátdehydrogenáza, kreatinkináza, amyláza, kyselina močová, amoniak, laktát) byly ve fyziologických mezích. Byly vyloučeny infekce hepatotropními viry, hepatitidy autoimunitního původu, deficit alfa-1-antitrypsinu, hypotyreóza, cystická fibróza a metabolické poruchy aminokyselin, sacharidů a glykogenu. Na rentgenu zápěstí kostní věk odpovídal věku dítěte a nebyly přítomny rentgenové známky rachitidy Na sonografickém vyšetření byla popsána hepatomegalie s vyšší echogenitou parenchymu a mírná splenomegalie. Pro podezření na střádavé onemocnění byla provedena biopsie jater. V hepatocytech byla popsána lyzozomálně lokalizovaná vakuolizace v hepatocytech, četné střádající makrofágy s velkým obsahem ceroidu a mírná fibróza portobilií. Patolog nález uzavřel jako střádavé onemocnění typu CESD. Pro potvrzení nebo vyloučení malabsorpčního syndromu byla provedena sukční enterobiopsie. Histologické vyšetření tenkého střeva včetně elektronové mikroskopie však neprokázalo žádné patologické změny. Provedené enzymatické vyšetření LKL v leukocytech potvrdilo těžký deficit na hodnotě 6% reziduální aktivity. Molekulárně genetickými metodami byla odhalena bodová mutace R44X a nová mutace Fl 18L.
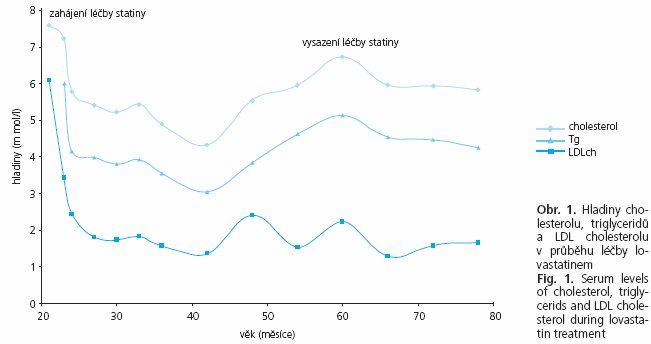
Léčba
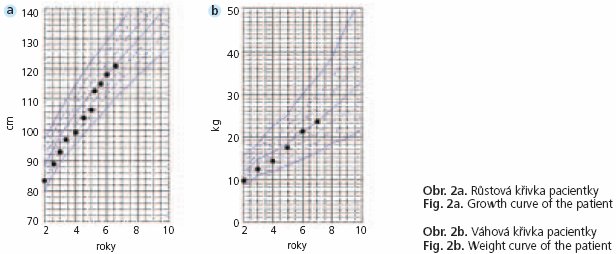
Od 2 let věku byla dietní léčba s omezením cholesterolu (200 mg/den) doplněna o podávání lovastatinu (20 mg/den). Při léčbě poklesla sérová hladina cholesterolu o 30 %, triglycerolů o 58 % a LDL cholesterolu o 36 % (obr. 1). Po přechodném vysazení lovastatinu došlo ke zvýšení hladin sledovaných tuků k původním hodnotám před léčbou. Hladina HDL cholesterolu se v průběhu léčby významně neměnila (0,52-0,67 mmol/1). Váha a výška pacientky se postupně upravily a po 5 letech léčby se pohybují kolem 50 percentilu (obr. 2). Velikost jater a ultrasonografický obraz se nezměnil. Trvají zvýšené sérové hladiny transamináz, které v průběhu sledování kolísaly (ALT 1,01-3,8 μkat/1; AST 1,23 až 3,14 μkat/1). Po celou dobu sledování pacientky byly hladiny kreatinkinázy vždy ve fyziologických mezích. Kontrolní jaterní biopsie ke zhodnocení histologického nálezu nebyla provedena. Léčba statiny probíhá bez komplikací a je dobře tolerována.
DISKUZE
Dosud bylo popsáno více než 50 případů pacientů s CESD. U většiny byla stanovena diagnóza před 20 rokem věku. Byly však popsány případy s velmi mírnou formou CESD, které byly diagnostikovány až po 40. roce věku(1,4). Je pravděpodobné, že mnozí pacienti s CESD jsou sledováni pod jinými diagnózami, jako např. heterozygotní forma familiární hypercholesterolémie, nealkoholická steatohepatitida nebo kryptogenní cirhóza. V České republice Elleder a kol. tuto variantu deficitu LKL diagnostikovali ve 13 případech v 11 rodinách. Pacienti byly rozděleni do 2 podskupin. Jedna skupina jako klinicky manifestní byla diagnostikována na základě hepatomegalie, která byla náhodně objevena při preventivních školních prohlídkách nebo při celkovém vyšetření při infekci dýchacích cest. Druhá skupina byla tvořena 3 dospělými pacienty ve věku 33, 41 a 53 let. Průběh nemoci byl benigní a klinické příznaky byly nespecifické(1). Variabilitu klinického obrazu ovlivňuje zbytková aktivita enzymu podobně jako u jiných střádavých nemocí(1,5).
V kazuistice je popsán nejmladší pacient s diagnostikovanou CESD v České republice. Klinicky kromě dominantní hepatomegalie byly příznaky neprospívání, které však nepatří do obrazu CESD. Byla popsána pouze jedna dospělá pacientka s CESD a s chronickým průjmem od dětství, úbytkem na váze, se sníženými hladinami vitaminů rozpustných v tucích a s histologickým obrazem střádání v lamina propria tenkého střeva(6). U naší pacientky histologické vyšetření střeva neodhalilo typický obraz popsaný u pacientů s CESD s přítomností střádajících makrofágů v lamina propria a tukových depozit v buňkách střevní mukózy a submukózy(7). Také nebyly přítomny změny na střevní sliznici, které by odpovídaly malabsorpčnímu syndromu. Příčina neprospívání zůstala neobjasněná, i když nelze vyloučit vliv salmonelové infekce, o jejímž průběhu a délce trvání nemáme dostatečné informace.
Hepatomegalie společně s mírně elevovanými transaminázami, hypercholesterolémií a nepřítomnou hypoglykémií by měla vést k podezření na CESD. Diagnóza je potvrzena stanovením zbytkové aktivity LKL v periferních leukocytech. V literatuře jsou popsány kazuistiky s vzácnou přechodnou variantou CESD. U pacientů s CESD byla popsána progresivní fibróza jater s portální hypertenzí a s gastrointestinálním krvácením, kterou bylo nutné řešit jaterní transplantací v prvém nebo ve druhém deceniu(8,9). Prenatální diagnostika je indikována u Wolmanovy nemoci a u přechodných forem CESD.
Léčba CESD je zaměřena na snížení hladiny cholesterolu jako rizikového faktoru pro rozvoj předčasné aterosklerózy a zastavení progrese jaterního onemocnění. Pozitivně je přijímaná léčba statiny která příznivě ovlivnila hladinu cholesterolu, a kromě toho nebyla zjištěna progrese nemoci při kontrolní jaterní biopsii. Vždy se však jednalo o pacienty s mírnou formou CESD(4). U pacientů se závažným průběhem CESD bylo popsáno snížení hladiny cholesterolu, ale nebyla ovlivněna progrese jaterního postižení(10), protože lovastatin neredukoval akumulaci tuků v hepatocytech(11). Jednalo se pouze o jednotlivá sdělení a vzhledem k malému počtu pacientů nebyla publikována žádná kontrolní studie léčby CESD statiny V jednotlivých případech byly použity statiny i v dětském věku u pacientů s CESD k prevenci rozvoje aterosklerózy Podání statinů bylo dobře tolerováno a nemělo žádné vedlejší účinky(11,12). U dětí a adolescentů s familiární hypercholesterolémií bylo v randomizovaných studiích z posledních let hodnoceno podání statinů jako bezpečné a účinné(13,14). Na podkladě literárních údajů a se souhlasem rodičů byla u naší pacientky zahájena léčba statiny, při které došlo ke snížení hladin tuků podobně jako u dvou popsaných pacientů dlouhodobě léčených lovastatinem(11). Hladiny transamináz po dobu sledování kolísaly a nelze vyloučit, že v souvislosti s léčbou. V průběhu podávání lovastatinu nebyly klinické ani biochemické známky myopatie nebo jiné závažné vedlejší účinky léčby. Nejasná zůstává prognóza jaterního postižení v dalším průběhu nemoci. Při progresi nemoci do cirhózy jater je indikována transplantace.
Prevalentní mutace u CESD je G934A sousedící se sestřihovým místem intronu 8, která způsobuje většinovou ztrátu exonu 8. Tato mutace je přítomna přibližně v 70 % všech mutantních chromosomů. u CESD(2). Pacienti bez této prevalentní mutace mají závažnější klinickou manifestaci nemoci(15) jako u popsané pacientky.
ZÁVĚR
CESD je velmi vzácné onemocnění, které patří do diferenciální diagnostiky hepatomegalie od kojeneckého věku až po dospělé pacienty. Léčba statiny je dobře tolerovaná, snižuje hladiny cholesterolu a triglyceridů, ale jejich vliv na progresi jaterního postižení je nejasný.
Poděkování patří dr. P. Lohsemu z Mnichova za molekulárně genetické určení mutací genu pro LKL a ing. H. Poupatové z Ústavu dědičných metabolických poruch v Praze za stanovení enzymové aktivity LKL v leukocytech.
LITERATURA
- 1. Elleder M, Poupětová H, Ledvinová J, et al. Deficit kyselé (lyzozomální) lipázy: přehled českých pacientů. Čas Lék čes 1999; 138: 719-724.
- 2. Lohse P, Maas S, Lose P, et al. Compound heterozygosity for a Wolman mutation is frequent aminy patiens with cholesterol ester storage disease. J Lipid Res 2000; 41: 23-31.
- 3. Glueck CJ, Lichtenstein P, Tracy T, et al. Safety ane efficacy of treatment of pediatrie cholesteryl ester storage disease with lovastatin. Pediatr Res 1992; 32: 559-565.
- 4. Gashe Ch, Aslanidis Ch, Kain R, et al. A novel variant of lysosomal acid lipase in cholesteryl storage disease associated with mild phenotype and improvement on lovastatin. J Hepatol 1997; 27: 744-750.
- 5. Hoeg JM, Demosky SJ, Pescovitz OH, et al. Cholesteryl ester storage disease and Wolman disease: phenotypic variants of lysosomal acid cholesteryl ester hydrolase deficiency. Am J Hum Genet 1984; 36: 1190-1203.
- 6. Drebber U, Andersen M, Rasper HU, et al. Severe chronic diarrhoea and wieght loss in cholesterol ester storage disease: a case report. World J Gastroenterol 2005; 11: 2364-2366.
- 7. Assmann G., Seedorf U. Acid lipase deficienty: Wolman disease and cholestery ester storage disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic and molecular bases of inhereted disease. New York: McGraw-Hill 1995: 2563-2587.
- 8. Leone L, Ippoliti PF, Antonicelli R, et al. Treatment and liver transplantation for cholesterol ester storage disease. J Pediatr 1995; 127: 509-510.
- 9. Di Bisceglie AM, Ishak KG, Rabin L, et al. Cholesteryl ester storage disease: hepathopatology and effects of therapy with lovastatin. Hepatology 1990; 11: 764-771.
- 10. Ferry GD, Whisennand HH, Finegold MJ, et al. Liver transplantation for cholesteryl ester storage disease. J Pediatr Gastroenterol 1991; 12: 376-378.
- 11. Rassoul F, Richter V, Lose P, et al. Long-term administration of the HMG-CoA reduktase inhibitor lovastatin in two patiens with cholesterol ester storage disease. Int J Clin Pharmacol Lher 2001; 39: 199-204
- 12. Tarantino MD, McNamara DJ, Grandstrom P, et al. Lovastatin therapy for cholesterol ester storage disease in two sisters. J Pediatr 1991; 118: 131-135.
- 13. McCrindle BW, Ose L, Marais AD. Efficacy and safety of otorvastatin in children and adolescents with familial hypercholesterolemia or severe hyperlipidemia: a multicenter, randomized, placebo-controlled trial. J Pediatr 2003; 142: 74-80.
- 14. Wiegman A, Hutten BA, de Groot E, et al. Efficacy and safety of statin therapy in children with familial hypercholesterolemia: a randomized controlled trial. JAMA 2004; 292: 331-337.
- 15. Anderson RA, Brusin GM, Parks JS. Lysosomal acid lipase mutations that determine phenotype in Wolman and cholesterol ester storage disease. Mol Gen Metab 1999; 68: 333-345.
Pro přístup k článku se, prosím, registrujte.
Výhody pro předplatitele
Výhody pro přihlášené