Gastrointestinálne prejavy familiárnej stredomorskej horúčky – problém aj v strednej Európe?
Zuzana Havlíčeková Orcid.org 1, Miloš Jeseňák Orcid.org 2, Nadežda Mišovicová3, Jana Kršiaková3, Ľubica Jakušová4, Peter Bánovčin Orcid.org 5
+ Pracoviště
Souhrn
Autoinflamačné (samozápalové) ochorenia sú skupina zriedkavých, geneticky determinovaných ochorení charakterizovaných spontánnou rekurentnou hyperaktiváciou mechanizmov vrodenej imunity pri absencii infekčných alebo autoimúnnych príčin. Familiárna stredomorská horúčka je najčastejšie monogénne autoinflamačné ochorenie, ktoré postihuje predovšetkým etnické skupiny žijúce v oblasti Stredomoria a Blízkeho východu. Výskyt v iných etnických skupinách je pomerne zriedkavý, ale vzhľadom k migrácii obyvateľstva sa s ním môžeme stretnúť aj v našich zemepisných šírkach. Popri febríliách sú bolesti brucha najčastejším klinickým príznakom familiárnej stredomorskej horúčky. Autori v článku prezentujú kazuistiky štyroch pediatrických pacientov sledovaných v Centre pre diagnostiku a liečbu primárnych imunodeficiencií JLF UK a UN Martin s gastrointestinálnou symptomatológiou, z ktorých len jeden mal predkov pochádzajúcich z oblasti Stredomoria.
Klíčová slova
bolesti brucha, děti, familiárna stredomorská horúčka, periodická horúčka
Autoinflamačné ochorenia
Autoinflamačné ochorenia (human autoinflammatory diseases) sú skupina zriedkavých, geneticky determinovaných ochorení charakterizovaných hyperaktiváciou mechanizmov vrodenej imunity pri absencii infekčných alebo autoimúnnych príčin. Väčšinou ide o monogénne ochorenia zapríčinené mutáciou génu kódujúceho proteín, ktorý zohráva kľúčovú úlohu v regulácii zápalovej odpovede. Ochorenia sú charakterizované rekurentnými, niekoľko dní až týždňov trvajúcimi epizódami febrílií spojených s eleváciou reaktantov akútnej fázy a variabilnými klinickými prejavmi, ktoré sa menia v závislosti od ochorenia (slizničné, kožné, serózne, osteoartikulárne) [1].
Familiárna stredomorská horúčka
Familiárna stredomorská horúčka (FMF – familial Mediterranean fever) je najčastejšie monogénne autoinflamatórne ochorenie, u ktorého sú bolesti brucha popri febríliách najčastejším klinickým syndrómom.
Kazuistika 1
V prvej kazuistike prezentujeme aktuálne takmer 4-ročného chlapca s anamnézou rekurentných febrílií nejasnej etiológie od 1,5 roka života, ktoré sa opakovali v 2–4týždňových intervaloch. Febrílie trvali okolo 3 dní, so slabou odpoveďou na antipyretiká, sprevádzané celkovou slabosťou, bolesťami brucha v oblasti epigastria, artralgiami a eleváciou nešpecifických zápalových parametrov. V medziobdobiach bol bez ťažkostí. Vzhľadom k podobnej klinickej symptomatológii u sesternice a balkánskemu pôvodu otca v diferenciálnej diagnostike zvažovaná FMF. Genetickým vyšetrením potvrdená prítomnosť patogénnej mutácie c.605G>A (p.Arg202Gln) MEFV v heterozygotnej forme. V 2 rokoch bola začatá profylakticky liečba kolchicínom. Na liečbe došlo k zmierneniu frekvencie a intenzity záchvatov. Medzi záchvatmi je hladina sérového amyloidu A (SSA) vo fyziologickom rozmedzí. Kazuistika prezentuje chlapca rizikového etnického pôvodu, s typickou klinickou symptomatológiou pri prítomnosti c.605G>A (p.Arg202Gln) mutácie MEFV v heterozygotnej forme. Táto mutácia je pomerne frekventná a predpokladá sa, že je spojená s vyšším rizikom amyloidózy [2].
Kazuistika 2
Aktuálne 5-ročné dieťa sledované gastroenterológom pre alergiu na obilniny. Na bezlepkovú diétu nastavené v 2. roku života na základe klinickej asociácie medzi bolesťami brucha, kašovitými stolicami a konzumáciou lepku. Na základe výsledkov laboratórnych a histologických vyšetrení celiakia vylúčená, stredne vysoko pozitívne protilátky proti omega-5-gliadínu. Na diéte došlo k zlepšeniu klinického stavu aj stavu výživy. Od 3. rokov sa objavujú febrílie s rekurenciou 4–6 týždňov, v trvaní 2–4 dni, sprevádzané dominantne bolesťou brucha, hnačkami, zvracaním, nechutenstvom a bolesťami kĺbov. Bolesti brucha s nechutenstvom sa začali objavovať aj mimo období záchvatov, dieťa začalo slabšie prospievať. Genetickým vyšetrením potvrdená patogénna mutácia c.2230G>T (p.Ala744Ser) MEFV v heterozygotnej forme. Dieťa je zároveň nosičom potenciálne patogénnej mutácie c.868G>A (p.Gln290Ter) v géne MVK pre enzým mevalonátkinázu. Na liečbe kolchicínom došlo k výraznému zmierneniu intenzity záchvatov. Pri nefrekventných záchvatoch sprevádzaných závažnou symptomatológiou podávaný s dobrým efektom rekombinantný analóg antagonistu receptora pre IL-1 (anakinra) niekoľko dní po sebe.
Kazuistika 3
Chlapec, 7 rokov, s rekurentnými febríliami od 5. roku života, opakujúcimi sa každých 4–6 týždňov, s bolesťami brucha, enteritídou, nauzeou a bolesťami kĺbov. Genetickým vyšetrením bola u pacienta potvrdená prítomnosť patogénnej mutácie (p.Lys695Arg) v 10. exéne génu MEFV v heterozygotnom stave a záro-veň v 2. exóne potvrdený variant p.Arg202Gln v heterozygotnom stave, ktorý je popísaný ako polymorfizmus asociovaný s ochorením (zložený heterozygot). Na liečbe kolchicínom má ťažkosti len sporadicky, s krátkym a miernym priebehom. Pri liečbe došlo k normalizácii C reaktivného proteinu ako aj sérového amyloidu. Aktuálne je vyšetrovaná tiež sestra pacienta s podobnou klinickou symptomatológiou.
Kazuistika 4
Dievča, 18 rokov, s recidívami febrílií spojených s bolesťou brucha, artralgiami a celkovou slabosťou od 3. roku. Genetickým vyšetrením až v 17. roku potvrdená FMF podmienená mutáciou p.Met694Val v MEFV géne (familial Mediterranean fever gene) v homozygotnej forme, ktorý je spojený s včasným nástupom ochorenia so závažnejším priebehom [3,4]. Odpoveď na liečbu kolchicínom aj anakinrou v záchvatovom období je len parciálna. Pretrváva mierne zvýšená hladina sérového amyloidu v medzizáchvatovom období, ktorá je známkou chronickej zápalovej odpovede spojenej so zvýšeným rizikom rozvoja orgánovej amyloidózy. Vzhľadom k tomu, že ani pri maximálnych terapeutických dávkach kolchicínu a podávaní anakinry v období záchvatov sa nedarí ochorenie adekvátne kontrolovať, plánujeme zahájenie liečby kanakinumabom (depotná monoklonová protilátka proti IL-1β).
Diskusia
FMF je najčastejšie monogénne auto-inflamačné ochorenie charakterizované self-limitujúcimi inflamatórnymi atakmi febrílií a polyserozitídy, s eleváciou nešpecifických zápalových parametrov. Najvýznamnejšou komplikáciou je amyloidóza, ktorá značne zvyšuje mortalitu ochorenia. K jej prevencii sa využíva liečba kolchicínom [5]. Postihuje predovšetkým etnické skupiny žijúce v oblasti Stredomoria (Sefardskí Židia, Arméni, Turci, Arabi, menej frekventne Gréci, Španieli a Taliani) s prevalenciou 1: 125–1 000. Výskyt v iných etnických skupinách je pomerne zriedkavý, ale vzhľadom k migrácii obyvateľstva sa s ním môžeme stretnúť aj v našich zemepisných šírkach [6,7]. V roku 2010 bola realizovaná nadnárodná štúdia mapujúca výskyt autoinflamatórnych ochorení v strednej a východnej Európe (vrátane Slovenskej aj Českej republiky), podľa ktorej bolo v tomto období v sledovanom regióne 11 pacientov s geneticky potvrdenou FMF a 49 predpokladaných ochorení, čo poskytuje výskyt v detskom veku 1: 465 500. Aj keď počet diagnostikovaných ochorení od tohto obdobia niekoľkonásobne vzrástol, výskyt ochorenia oproti krajinám západnej Európy je stále nižší a mnohé prípady ostávajú nediagnostikované [8].
Ochorenie je geneticky podmienené. MEFV gén je lokalizovaný na 16. chromozóme a kóduje proteín pyrín. Pyrín sa nachádza dominantne v neutrofiloch, makrofágoch. Zohráva kľúčovú úlohu v procesoch vrodenej imunity pri regulácii apoptózy, zápalových reakcií a produkcii viacerých cytokínov. Za fyziologických podmienok aktivuje NF-κβ a prokaspázu-1, čím dochádza k supresii produkcie IL-10 a tvorbe aktívneho IL-1β. Prítomnosť patologickej mutácie vedie k prehnaným zápalovým odpovediam s excesívnou produkciou IL-1β a následne proteínov akútnej fázy, vrátane SSA [9]. Doposiaľ bolo popísaných viac ako 300 genetických polymorfizmov MEFV. Väčšina polymorfizmov je zriedkavých, bez jednoznačného klinického významu [10]. Klasická dedičnosť ochorenia je autozomálne recesívna, avšak fenotypové prejavy sú vyjadrené asi u 1/4 heterozygotov. Hovoríme o tzv. autozomálne dominantnej dedičnosti s nízkou penetranciou (event. pseudodominatnej dedičnosti) [4]. U heterozygotov s vyjadrenými klinickými prejavmi sa v etiopatogenéze zvažuje účasť epigenetických mechanizmov. Fenotypové prejavy ochorenia sú tiež ovplyvnené rôznymi environmentálnymi faktormi.
FMF je charakterizovaná krátkymi febrilnými epizódami trvajúcimi obvykle 1–4 dni, ktoré sú spojené s prejavmi serozitídy (peritonitída, pleuritída, perikarditída, synovitída) a kožnými prejavmi [11]. Popri febríliách sú bolesti brucha najčastejším klinickým FMF, nasledované kĺbnymi ťažkosťami, ktoré postihujú predovšetkým jednotlivo veľké kĺby na dolných končatinách a môžu trvať dlhšie ako iné symptómy [12]. V zriedkavých prípadoch boli popísané aj ochorenia s afebrilným priebehom [13]. Pleuritída alebo perikarditída podmieňujú bolesti na hrudníku. Prejavy pleuritídy sú väčšinou jednostranné, trvajú 1–4 dni a sú spojené s tvorbou výpotku detekovateľného sonograficky. Prejavy perikarditídy môžu byť prolongovanejšie a väčšinou ustupujú do 14 dní [14]. Ďalšie prejavy zahŕňajú kožné (najčastejšie eryzipeloidný erytém), myalgie (asociované s fyzickou námahou, febrilné), u mužov môže dôjsť k rozvoju prejav imitujúcich akútne skrótum. V rámci diferenciálnej diagnostiky bolestí hlavy je ako príčina zvažovaná rekurentná aseptická meningitída asociovaná s FMF [15]. Popri charakteristicky vyskytujúcom sa obličkovom poškodení spojenou s ukladaním amyloidu (uvedené v kapitole komplikácie) sa u pacientov s FMF môžeme zriedkavo stretnúť s rôznymi prejavmi renálneho poškodenia bez prítomnosti amyloidózy (proteinúria, hematúria, rekurentná akútna pyelonefritída a glomerulonefritída) [11].
Ataky môžu mať prodromálnu symptomatológiu, ktorá sa objavuje 1–2 dni vopred a zahŕňa dyskomfort, rôzne fyzické a emočné problémy [16]. Môžu byť trigerované emočným stresom, fyzickou aktivitou, infekčným ochorením, chirurgickým výkonom, nástupom menštruácie, ale väčšinou nie je možné identifikovať špecifický vyvolávajúci činiteľ. Sú nepravidelné, väčšinou krátkeho trvania (menej ako 3 dni) a spontánne ustúpia. Majú väčšinou podobnú klinickú symptomatológiu väčšinou sa opakujúcu viac krát ročne. Ale pozorovaná bola rôzna variabilita asymptomatických intervalov od týždňov až dokonca po roky. Medzi atakmi ochorenia sú pacienti zvyčajne asymptomatickí, s normalizáciou proteínov akútnej fázy. Štúdie však naznačili, že až do 2/3 pacientov môže mať aj v asymptomatickom období laboratórne prejavy aktivácie zápalovej kaskády [17]. Prvé prejavy ochorenia väčšinou prichádzajú pred 10. rokom života, z toho u 1/3 sa prejavy objavujú včasne, už pred 2. rokom. Po 20. roku sa prejaví okolo 20 % prípadov. U detí s včasným nástupom sa stretávame s výraznejšími prejavmi, ktorých intenzita s vekom klesá [1]. Vo včasnom veku sa môže ochorenie prejavovať len izolovanými febríliami a symptomatológia asociovaná so sérozitídou sa vyvinie až neskôr. Len zriedkavo sú prejavy chronického, nie epizodického charakteru (protrahované myalgie s febrilným priebehom, FMF-asociovaná vaskulitída, perzistencia elevácie zápalovej aktivity bez jednoznačnej klinickej symptomatológie).
Amyloid môže vytvárať depozity vo viacerých orgánoch a pri dlhodobom, neliečenom ochorení spôsobovať poruchu ich funkcie (obličky, nadobličky, črevo, slezina, pečeň, žalúdok, štítna žľaza, srdce a pľúca). Pri neliečenom ochorení je riziko amyloidózy okolo 60–75 %. Progresívne postihnutie obličiek, ktoré sa vyskytuje najfrekventnejšie, môže vyústiť až do chronického obličkového zlyhávania je najobávanejšou komplikáciou [11,18].
S FMF bol popísaný výskyt asociovaných ochorení – vaskulitídy (Henoch-Schönleinova purpura, polyarteritis nodosa), aterosklerotické zmeny, Behcetova choroba, ale aj nešpecifické črevné zápalové ochorenia (IBD – inflammatory bowel disease) [19–21].
Gastrointestinálne prejavy familárnej stredomorskej horúčky
Gastrointestinálna symptomatológia je spolu s febríliami najčastejším klinickým prejavom a vyskytuje sa až u 95 % pacientov [22–25]. Prejavy môžeme rozdeliť do niekoľkých skupín:
1. Peritonitída a asociované prejavy, ktorá je súčasťou febrilného ataku a môže prebiehať pod obrazom akútnej peritonitídy a črevnej obštrukcie. Zápal peritonea je najčastejšou príčinou bolestí u pacientov s FMF. Úvodne je bolesť rôzne lokalizovaná, variabilnej intenzity, bez typickej predominancie, s rýchlou generalizáciou. Pri klinickom vyšetrení nachádzame typické známky asociované s peritoneálnym dráždením, laboratórne eleváciu nešpecifických zápalových parametrov a na rentgenovom snímke obraz subileózneho až ileózneho stavu, sonograficky je možno potvrdiť tekutinu v dutine brušnej. Aj keď pre väčšinu pacientov je typická porucha pasáže gastrointestinálnym traktom (GIT) spojená s rozvojom ileózneho stavu, 10 až 20 %, predovšetkým deti, môže mať hnačky [23]. Vzhľadom ku klinickému obrazu tzv. akútneho brucha je u niektorých pacientov indikovaná exploratívna laparotómia, najčastejšie pre podozrenie na akútnu apendicitídu. Peroperačný nález je väčšinou bez patológie, len s malým množstvom sterilnej, zápalovej peritoneálnej tekutiny. Podľa retrospektívnej štúdie Kaşifoğlu et al 90 % operačných výkonov prebehne pred stanovením diagnózy, ale zvyšných 10 % je realizovaných pri známej diagnóze FMF, väčšinou pre ileózny stav [26]. Ústup bolestí brucha sa začína za 6–12 hod a zvyčajne trvajú 24–48 hod [22,24,25]. Následkom opakovaných peritonitíd môže u niektorých pacientov dochádzať k akumulácii tekutiny v peritoneálnej dutine, ktorá je bohatá na fibrín, polymorfonukleárne leukocyty a má charakter exsudátu. Opakovaná akumulácia exsudátu môže zapríčiniť tvorbu adhézií peritonea a viesť ku komplikáciám (viscerálne adhézie, mechanická črevná obštrukcia, volvulus, strangulácia čreva) [25]. Prejavy mechanického ilea sú u pacientov s FMF v porovnaní s bežnou populáciou frekventnejšie [27]. Vyskytujú sa asi u 3 % prípadov FMF bez anamnézy predchádzajúceho chirurgického výkonu [26]. Ďalšou, ale zriedkavou komplikáciou rekurentných atakov peritonitídy je rozvoj sklerotizujúcej enkapsulovanej peritonitídy [22]. V minulosti niektoré centrá odporúčali u pacientov s FMF realizovať,,preventívnu apendektómiu“ aby sa zabránilo diagnostickým pochybnostiam pri atakoch bolestí brucha spojených s peritonitídou. Aktuálne sa tento postup nepovažuje sa vhodný, aj vzhľadom k riziku peritoneálnych adhézií s ich komplikáciami.
2. Ochorenia, ktoré prebiehajú pod obrazom funkčných ochorení GIT, sú asociované s FMF približne u 5 % pacientov a môžu byť spojené s bolesťami brucha, zmenami frekvencie stolice nezávislými od atakov [28]. Brik et al poukázali na 20% výskyt mutácie MEFV v homozygotnej forme v skupine 59 Sefardských Židov v detskom veku, ktorí boli na základe dostupných výsledkov vyšetrení sledovaní pre funkčné bolesti brucha. Autori preto uzatvárajú, že v diferenciálnej diagnostike rekurentných bolestí brucha by v stredomorských oblastiach mala byť zvážená FMF [29].
3. Klinické prejavy asociované s amyloidózou – mikroskopické depozity amyloidu v stene GIT – je možné nájsť už vo včasných fázach ochorenia. Amyloidóza čreva sa stáva symptomatickou len u nízkeho percenta pacientov, po dlhoročnom ukladaní depozitov amyloidu, pri postihnutí celej hrúbky steny čreva. Klinické prejavy sú variabilné (zápcha, protrahovaná hnačka, závažná malabsorbcia, predovšetkým pri prejavoch bakteriálneho prerastania a dekonjugácie žlčových kyselín následkom zníženej motility tenkého čreva). Niekedy môže byť problematické odlíšiť hnačku, ktorá je asociovaná s liečbou kolchicínom [28]. Ďalšími komplikáciami amyloidózy GIT je ischemická enterokolitída, ktorá môže vyústiť do fibrózy, slizničných ulcerácií, črevnej obštrukcie alebo perforácie. Klinické, rádiologické a histopatologické nálezy amyloid-asociovanej ischemickej kolitídy môžu imitovať Crohnovu chorobu (CD – Crohn’s disease). Významným rozdielom je absencia transmurálnach lymfoidných agregátov a granulómov.
4. Gastrointestinálne slizničné postihnutie bez amyloidózy (kolitída, aftózna gastroduodenitída, perianálne posti-hnutie) [30].
5. Hepatobiliárne prejavy – okrem hepatomegálie (5 %) sa môžeme stretnúť s idiopatickou cirhózou pečene, ktorá sa u pacientov s FMF vyskytuje v porovnaní s bežnou populáciou frekventnejšie a predpokladá sa zatiaľ neobjasnená asociácia medzi ochoreniami [31–33]. Podobne je pozorovaný častejší výskyt nealkoholovej steatohepatitídy, pravdepodobne následkom pôsobenia proinflamatórnych cytokínov a mutácie MEFV génu na pečeň. Zvažuje sa, že postupný vývoj ochorenia cez nealkoholovú steatohepatitídu až po cirhózu pečene môže byť príčinou vyššieho výskytu cirhózy pečene u pacientov s FMF [34]. Niektoré práce upozornili na vyšší výskyt cholecystolitiázy [22].
Asociácia FMF s IBD
Klinická štúdia Cattan et al poukázala na 5-násobne vyšší výskyt IBD v populácii non-Aškenázskych Židov v porovnaní s bežnou populáciou. Až u 1/2 pacientov išlo o ochorenia so závažným priebehom [20]. Podobne Fidder et al potvrdili vyšší výskyt CD u pacientov s FMF. CD sa u nich prejavila skôr vo vyššom veku (40 vs. 26 rokov), s porovnateľnými klinickými prejavmi ako u kontrolnej skupiny, avšak s vyšším počtom atakov FMF a vyšším rizikom sekundárnej amyloidózy [21]. Vyšší výskyt IBD v populácii pediatrických pacientov s FMF potvrdili u tureckých detí Beşer et al. Z 78 detí malo asociované IBD až 14,8 % (11 s ulceróznou kolitídou – UC, 1 CD), väčšinou stredne závažného priebehu [35]. Uslu et al hodnotili prítomnosť polymorfizmov MEFV génu u 33 tureckých detí s IBD (14 CD, 16 UC, 3 indeterminovaná kolitída – IC). Mutácia MEFV bola detegovaná u 1/4 alel. FMF bola diagnostikovaná u 21,2 % pacientov (4 CD, 2 UC, 1 IC). Najfrekventnejšia bola mutácia M694V [36]. Asociácia ochorení je vysvetľovaná možným vzájomným ovplyvňovaním génových produktov NOD2 a MEFV, ktoré sa zúčastňujú mechanizmov bunkovej apoptózy, produkcie cytokínov a regluácie zápalových procesov. Oba gény sú lokalizované na 16. chromozóme a ich proteínové produkty majú podobnú štruktúru.
Diagnostika
Diagnostika FMF je postavená na prítomnosti typickej klinickej symptomatológie, podporenej etnickým pôvodom a rodinnou anamnézou. Neexistuje žiadny špecifický marker pre FMF. V priebehu akútnych atakov ochorenia nachádzame leukocytózu, eleváciu reaktantov akútnej fázy vrátane SSA. Medzi záchvatmi dochádza typicky k ich normalizácii, aj keď v zriedkavých prípadoch závažného, nekontrolovaného ochorenia môžu pretrvávať medzi záchvatmi mierne zvýšené hladiny nešpecifickej zápalovej aktivity. Prejavy chronickej, silentnej inflamácie spojenej s perzistenciou elevácie SSA nachádzame u 30–60 % pacientov. Porucha obličkových funkcií s proteínúriou vzniká ako následok amyloidózy pri dlhodobom neliečenom ochorení [17].
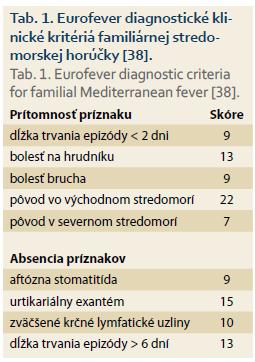
Pri diagnostike v regiónoch s typickým výskytom FMF sa využívajú kritériá podľa Tel-Hashomera s vysokou senzitivitou a špecificitou, ktorá dosahuje až 95, resp. 97 %, ktoré boli neskôr zjednodušené Livnehom et al [37]. Pre pediatrickú populáciu boli modifikované kritériá podľa Yalcinkaya-Ozena dosahujúce senzitivitu okolo 87 % [38]. Pre krajiny s nízkym výskytom FMF však nie sú žiadne z nich aplikovateľné pre nízku špecificitu. V roku 2015 boli publikované Eurofever klinické diagnostické kritériá so 68% senzitivitou a 87% špecificitou, ktoré je možné aplikovať aj v našich podmienkach. Podľa nich by mala byť diagnóza FMF zvážená, pokiaľ pacient dosiahne minimálne 60 bodov (tab. 1) [39].
Genetické vyšetrenie
Genetické vyšetrenie má význam pri podpore diagnózy FMF, nie však pri jej vylúčení. Pozitívna prediktívna hodnota vyšetrenia je okolo 70–80 %. Podľa aktuálneho konsenzu o genetickom vyšetrení by sa pri diagnostike FMF malo vyšetrovať 14 polymorfizmov MEFV génu, z ktorých 9 je jednoznačne patogénnych (E167D, T267I, M694V, V726A, M680I, M694I, R761H,A744S, I692del) a ostatných 5 variánt je zatiaľ bez jednoznačnej klinickej významnosti [10]. Ako už bolo spomínané pri etiopatogenéze ochorenia, ochorenie je dedičné autozomálne recesívne, ale klinické prejavy má vyjadrené asi 1/4 heterozygotov. Zvažovaný je tzv. efekt dávky odlišujúci mutácie s nízkou a vysokou penetranciou, ktoré sa diferencujú závažnosťou klinickej symptomatológie. Polymorfizmus M694V v homozygotnej forme alebo pri kombinovanej heterozygotnej forme je spojený s včasným nástupom ochorenia so závažnejším priebehom [3,4]. Aj pacientov s typickou symptomatológiou s jednou mutáciou alebo bez potvrdenej mutácie by mal byť zvážený diagnostický kolchicínový test na 3–6 mesiacov.
Liečba
Základom liečby FMF je dlhodobé podávanie kolchicínu, ktorý umožňuje prevenciu záchvatov ochorenia, kontroluje subklinické zápalové prejavy u väčšiny pacientov a z dlhodobého hľadiska znižuje riziko amyloidózy. Dávka pre deti je 0,5–1 mg/deň, pre dospelých 1– 2 mg/deň. Antiinflamatórny efekt kolchicínu je zabezpečený prostredníctvom prevencie aktivácie neutrofilov tvorbou β-tubulín-kolchicínových komplexov, inhibície oligomerizácie pyrínu, inhibície expresie kaspázy-1 a syntézy TNF-α. Liečba kolchicínom je bezpečná, je však pravidelne potrebné monitorovať možné nežiaduce účinky. Najčastejším nežiaducim efektom liečby sú gastrointestinálne prejavy spojené s hnačkou a laktózovou intoleranciou. Medzi zriedkavejšie patrí deficit vitamínu B12, reverzibilná periférna neuritída, myopatia, myelosupresia a alopécia. Efektivita liečby môže byť hodnotená sledovaním hladiny SSA. Ak je táto liečba tolerovaná, je aplikovaná celoživotne bez prerušovania. U pacientov, ktorí liečbu netolerujú alebo je neúčinná (5–10 %), sa zdá byť prínosom podávanie preparátov biologickej liečby – antagonistov IL-1 (napr. anakinra, kanakinumab, rilonacept) [40].
Záver
Napriek tomu, že FMF sa v strednej Európe vyskytuje zriedkavo, mala by byť zvážená v diferenciálnej diagnostike u detí s rekurentnými bolesťami brucha sprevádzanými febríliami, s eleváciou nešepecifických zápalových parametrov. Obzvlášť dôležitým údajom je opakovaný nález voľnej tekutiny v brušnej dutine pri záchvate teploty a pri elevácii zápalových markerov. Aktuálne štúdie upozornili aj na to, že až 1/2 pacientov pred stanovením diagnózy FMF absolvovala gastroenterologické vyšetrenie pre bolesti brucha alebo iné gastrointestinálne prejavy, ale vo väčšine prípadov neprispelo k správnemu stanoveniu diagnózy.
Táto práca bola podporená projektom Centrum experimentálnej a klinickej respirológie II. (ITMS 26220120034), spolufinancovaným z projektov EÚ.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
The Editorial Board declares that the
manuscript met the ICMJE „uniform requirements“ for biomedical papers.
Doručeno/Submitted: 14. 9. 2017
Přijato/Accepted: 25. 11. 2017
doc. MUDr. Miloš Jeseňák, PhD., MBA
Klinika detí a dorastu
JLF UK a UN Martin
Kollárova 2
036 59 Martin
jesenak@gmail.com
Pro přístup k článku se, prosím, registrujte.
Výhody pro předplatitele
Výhody pro přihlášené
Literatura
1. Cush JJ. Autoinflammatory syndromes. Dermatol Clin 2013; 31 (3): 471–480. doi: 10.1016/j.det.2013.05.001.
2. Nursal AF, Tekcan A, Kaya SU et al. Mutational spectrum of the MEFV gene in AA amyloidosis associated with familial Mediterranean fever. Iran J Kidney Dis 2016; 10 (3): 107–112.
3. Ozturk C, Halicioglu O, Coker I et al. Association of clinical and genetical features in FMF with focus on MEFV strip assay sensitivity in 452 children from western Anatolia, Turkey. Clin Rheumatol 2012; 31 (3): 493–501. doi: 10.1007/s10067-011-1876-1.
4. Marek-Yagel D, Berkun Y, Padeh S et al. Clinical disease among patients heterozygous for familial Mediterranean fever. Arthritis Rheum 2009; 60 (6): 1862–1866. doi: 10.1002/art.24570.
5. Fonnesu C, Cerquaglia C, Giovinale G et al. Familial Mediterranean fever: a review for clinical management. Joint Bone Spine 2009; 76 (3): 227–233. doi: 10.1016/j.jbspin.2008.08.004.
6. Kisacik B, Yildirim B, Tasliyurt T et al. Increased frequency of familial Mediterranean fever in northern Turkey: a population-based study. Rheumatol Int 2009; 29 (11): 1307–1309. doi: 10.1007/s00296-009-0849-z.
7. Ben-Chetrit E, Touitou I. Familial Mediterranean fever in the world. Arthritis Rheum 2009; 61 (10): 1447–1453. doi: 10.1002/art.24458.
8. Toplak N, Dolezalovà P, Constantin T et al. Periodic fever syndromes in Eastern and Central European countries: results of a pediatric multinational survey. Pediatr Rheumatol Online J 2010; 8: 29. doi: 10.1186/1546-0096-8-29.
9. Ozkurede VU, Franchi L. Immunology in clinic review series; focus on autoinflammatory dis-eases: role of inflammasomes in autoinflammatory syndromes. Clin Exp Immunol 2012; 167 (3): 382–390. doi: 10.1111/j.1365-2249.2011.04535.x.
10. Shinar Y, Obici L, Aksentijevich I et al. Guidelines for the genetic diagnosis of hereditary recurrent fevers. Ann Rheum Dis 2012; 71 (10): 1599–1605. doi: 10.1136/annrheumdis-2011-201271.
11. Alghamdi M. Familial Mediterranean fever, review of the literature. Clin Rheumatol 2017; 36 (8): 1707–1713. doi: 10.1007/s10067-017-3715-5.
12. Lidar M, Kedem R, Mor A et al. Arthritis as the sole episodic manifestation of familial Mediterranean fever. J Rheumatol 2005; 32 (5): 859–862.
13. Vogel Y, Büchner NJ, Haverkamp T et al. Familial Mediterranean fever. Rare manifestation without fever and with inconspicuous family case history. Dtsch Med Wochenschr 2008; 133 (31–32): 1621–1624. doi: 10.1055/s-0028-1082777.
14. Kees S, Langevitz P, Zemer D et al. Attacks of pericarditis as a manifestation of familial Mediterranean fever (FMF). QJM 1997; 90 (10): 643–647.
15. Capron J, Grateau G, Steichen O. Is recurrent aseptic meningitis a manifestation of familial Mediterranean fever? A systematic review. Clin Exp Rheumatol 2013; 31 (3 Suppl 7): 127–132.
16. Lidar M, Yaqubov M, Zaks N et al. The prodrome: a prominent yet overlooked pre-attack manifestation of familial Mediterranean fever. J Rheumatol 2006; 33 (6): 1089–1109.
17. Lachmann HJ, Sengül B, Yavuzşen TU et al. Clinical and subclinical inflammation in patients with familial Mediterranean fever and in heterozygous carriers of MEFV mutations. Rheumatology (Oxford) 2006; 45 (6): 746–750. doi: 10.1093/rheumatology/kei279.
18. Livneh A, Langevitz P, Shinar Y et al. MEFV mutation analysis in patients suffering from amyloidosis of familial Mediterranean fever. Amyloid 1999; 6 (1): 1–6.
19. Rigante D, Lopalco G, Tarantino et al. Non-canonical manifestations of familial Mediterranean fever: a changing paradigm. Clin Rheumatol 2015; 34 (9): 1503–1511. doi: 10.1007/s10067-015-2916-z.
20. Cattan D, Notarnicola C, Molinari N et al. Inflammatory bowel disease in non-Ashkenazi Jews with familial Mediterranean fever. Lancet 2000; 355 (9201): 378–379.
21. Fidder HH, Chowers Y, Lidar M et al. Crohn disease in patients with familial Mediterranean fever. Medicine (Baltimore) 2002; 81 (6): 411–416.
22. Livneh A, Langevitz P, Zemer D et al. The changing face of familial Mediterranean fever. Semin Arthritis Rheum 1996; 26 (3): 612–627.
23. Onen F. Familial Mediterranean fever. Rheumatol Int 2006; 26 (6): 489–496. doi: 10.1007/ s00296-005-0074-3.
24. Tunca M, Akar S, Onen F et al. Familial Mediterranean fever (FMF) in Turkey: results of a of a nationwide multicenter study. Medicine (Baltimore) 2005; 84 (1): 1–11.
25. Sohar E, Gafni J, Pras M et al. Familial Mediterranean fever. A survey of 470 cases and review of the literature. Am J Med 1967; 43 (2): 227–253.
26. Kaşifoğlu T, Cansu DU, Korkmaz C. Frequency of abdominal surgery in patients with familial Mediterranean fever. Intern Med 2009; 48 (7): 523–526.
27. Berkun Y, Ben-Chetrit E, Klar A. Peritoneal adhesions and intestinal obstructions in patients with familial Mediterranean fever – are they more frequent? Semin Arthritis Rheum 2007; 36 (5): 316–321. doi: 10.1016/j.semarthrit.2006.11.002.
28. Mor A, Gal R, Livneh A. Abdominal and digestive system associations of familial Mediterranean fever. Am J Gastroenterol 2003; 98 (12): 2594– 2604. doi: 10.1111/j.1572-0241.2003.08784.x.
29. Brik R, Litmanovitz D, Berkowitz D et al. Incidence of familial Mediterranean fever (FMF) mutations among children of Mediterranean extraction with functional abdominal pain. J Pediatr 2001; 138 (5): 759–762. doi: 10.1067/mpd.2001.113357.
30. Gurkan OE, Dalgic B. Gastrointestinalmucosal involvement without amyloidosis in children with familial Mediterranean fever. J Pediatr Gastroenterol Nutr 2013; 57 (3): 319–323. doi: 10.1097/MPG.0b013e318295fc65.
31. Aharoni D, Hiller N, Hadas-Halpern I. Familial Mediterranean fever: abdominal imaging findings in 139 patients and review of the literature. Abdom Imaging 2000; 25 (3): 297–300.
32. Ishak GE, Khoury NJ, Birjawi GA et al. Imaging findings of familial Mediterranean fever. Clin Imaging 2006; 30 (3): 153–159. doi: 10.1016/j.clinimag.2005.07.002.
33. Tweezer-Zaks N, Doron-Libner A, Weiss P et al. Familial Mediterranean fever and cryptogenic cirrhosis. Medicine (Baltimore) 2007; 86 (6): 355–362. doi: 10.1097/MD.0b013e31815be056.
34. Rimar D, Rosner I, Rozenbaum M et al. Familial Mediterranean fever: an association with non-alcoholic fatty liver disease. Clin Rheumatol 2011; 30 (7): 987–991. doi: 10.1007/ s10067-011-1718-1.
35. Beşer OF, Kasapçopur O, Cokuğraş FC et al. Association of inflammatorybowel disease with familial Mediterranean fever in Turkish children. J Pediatr Gastroenterol Nutr 2013; 56 (5): 498–502. doi: 10.1097/MPG.0b013e31827dd763.
36. Uslu N, Yüce A, Demir H et al. The association of inflammatory bowel disease and Mediterranean fever gene (MEFV) mutations in Turkish children. Dig Dis Sci 2010; 55 (12): 3488–3494. doi: 10.1007/s10620-010-1178-5.
37. Livneh A, Langevitz P, Zemer D et al. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum 1997; 40 (10): 1879–1885.
38. Demirkaya E, Saglam C, Turker T et al. Performance of different diagnostic criteria for familial Mediterranean fever in children with periodic fevers: results from a multicenter international registry. J Rheumatol 2006; 43 (1): 154–160. doi: 10.3899/jrheum.141249.
39. Federici S, Sormani MP, Ozen S el. al. Evidence-based provisional clinical classification criteria for autoinflammatory periodic fevers. Ann Rheum Dis 2015; 74 (5): 799–805. doi: 10.1136/annrheumdis-2014-206580.
40. Ben-Chetrit E, Ozdogan H. Non-response to colchicine in FMF – definition, causes and suggested solutions. Clin Exp Rheumatol 2008; 26 (4 Suppl 50): S49–S51.