
Keywords
pharmacotherapy
pharmacodynamics
therapeutic targets
Abstract
Research and development of new drugs for the treatment of inflammatory bowel diseases focuses on approximately 20 potential pharmacodynamic targets. Most of these are receptors for inflammatory metors and enzymes involved in their function, as well as cytokines themselves. In addition, influencing micro-RNA, immune checkpoints, or an inflammasome appears to be a potential therapeutic option. The most advanced developments to date are in the areas of TL1A cytokine inhibition, microRNA-124 function stimulation, interleukin 6 trans-signalling pathway inhibition, and the type I transforming growth factor β receptor blockade. However, none of the tested targets appear to be entirely specific to the pathophysiology of inflammatory bowel diseases, which is a prerequisite for the development of potentially curative pharmacotherapy.
Úvod
Idiopatické střevní záněty (IBD) jsou skupinou chronických autoimunitních zánětlivých onemocnění, postihující zejména, ale nejenom gastrointestinální trakt. Jsou to především ulcerózní kolitida (UC) a Crohnova choroba (CD), které se od sebe v řadě charakteristik liší, nicméně mají společnou řadu patofyziologických mechanizmů, díky kterým je i v jejich léčbě řada společných postupů. Průběh onemocnění je charakterizován střídáním různě dlouhých období remise a relapsů. Etiologie je neznámá a patogeneze pouze částečně. Nejpravděpodobněji v ní hraje roli interakce faktorů zevního prostředí a mikrobiomu vedoucí u geneticky predisponovaných jedinců k abnormální aktivaci imunitního systému ve sliznici gastrointestinálního traktu, kde prostřednictvím lokálně uvolňovaných prozánětlivých cytokinů dochází ke vzniku zánětu a destrukci tkání. V dospělé populaci rozvinutých zemí je incidence IBD stabilní, zatímco v dětské populaci narůstá. Jejich prevalence se zvyšuje ve všech věkových skupinách stejně [1].
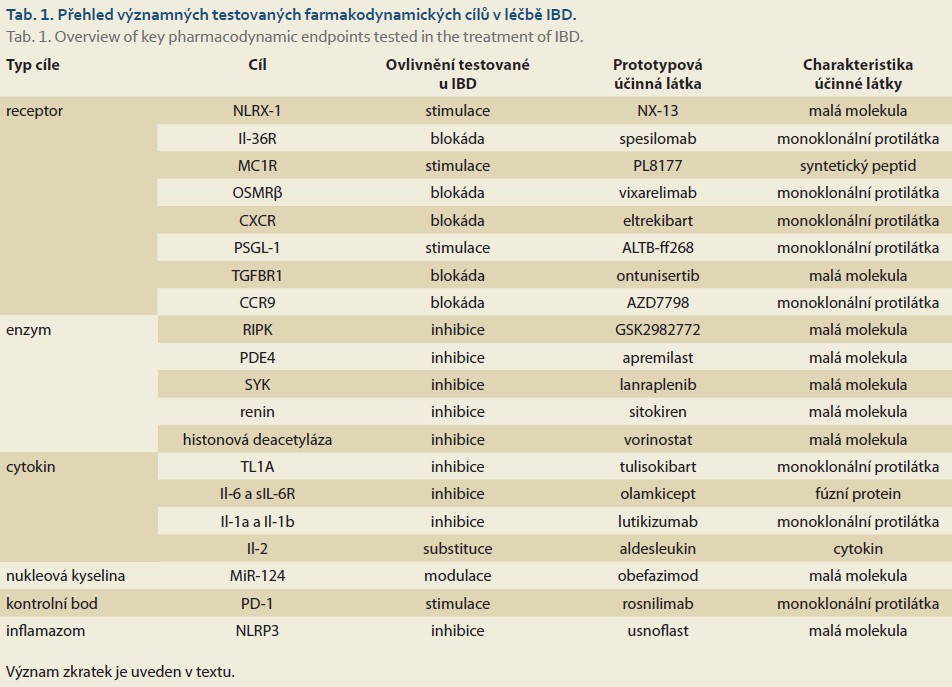
Současné možnosti farmakoterapie IBD zahrnují několik tříd léků, z těch klasických jsou to zejména glukokortikoidy, aminosalicyláty a imunosupresiva, v určitých situacích i antibiotika. Zásadní pokrok přinesla moderní cílená léčba zahrnující biologické léky ze skupiny monoklonálních protilátek i malé molekuly, především JAK inhibitory a S1P modulátory. Z monoklonálních protilátek jsou dnes již běžně používána léčiva cílená proti TNFα, integrinům (především α4β7) a interleukinu 23. Farmakologickou léčbu doplňují také dieta, probiotika a enterální i parenterální výživa. Navzdory dnes již širokým možnostem farmakologické terapie vykazuje významná část pacientů primární nebo sekundární rezistenci k dostupné léčbě. Z klinických dat je zřejmé, že i nová léčiva jsou v monoterapii účinná u maximálně 60 % pacientů [2]. Tato skutečnost dává impulz k výzkumu nových terapeutických cílů a vývoji na ně cílených léčivých přípravků. Dále je uveden přehled těchto potenciálních farmakodynamických cílů a možnosti jejich farmakologického ovlivnění. Přehled je uveden v tab. 1.
Inhibice cytokinu TL1A
TL1A (tumor necrosis factor-like cytokine 1A) je cytokin, který je členem superrodiny TNF. Jinak se také nazývá TNF superfamily member 15 (TNFSF15) nebo vascular endothelial growth inhibitor (VEGI). V lidském organizmu se vyskytuje membránově vázaný i volný. Je exprimován slizničními antigen prezentujícími buňkami (makrofágy, monocyty, dendritické buňky, endotelie a T-lymfocyty, zejména Th1 a Th17) po antigenní stimulaci. Jeho receptorem je DR3 (death domain receptor 3) nacházející se zejména na T-lymfocytech, více na CD4+ než CD8+. Vazba TL1A na jeho receptor v souhře s dalšími imunologickými podněty vede ke stimulaci T-lymfocytů, k proliferaci a produkci cytokinů a zesílení Th1, Th2 a Th17 imunitní odpovědi. Samotný vliv TL1A obvykle pro diferenciaci naivních T buněk do linií Th1, Th2 a Th7 nestačí, ale funguje v synergii s jinými stimuly [3,4]. Prostřednictvím IL-12 a IL-23 zvyšuje produkci INFγ z Th1 buněk a produkci IL-17 u Th17 buněk. Výše exprese TL1A u IBD koreluje s tíží onemocnění a zdá se, že prostřednictvím aktivace fibroblastů se také účastní dějů zodpovědných za rozvoj fibrózy v gastrointestinálním traktu [5].
Prototypovou látkou TL1A inhibitorů je tulisokibart. Jedná se o humanizovanou monoklonální protilátku IgG1 κ (kappa) proti volnému i membránově vázanému TL1A. Zabraňuje vazbě TL1A na receptor DR3, čímž potlačuje aktivaci zánětlivých drah, včetně Th1/Th7 signálních drah, zvyšuje aktivitu regulačních T-buněk a tlumí profibrotické procesy [6]. Tulisokibart má ukončeny dvě klinické studie fáze II pro UC i CD. Ve studii ARTEMIS-UC po 12 týdnech užívání tulisokibartu dosáhlo klinické remise 26 %, endoskopického zlepšení 37 % a klinické odpovědi 66 % zúčastněných se střední až těžkou aktivní UC. Pro CD proběhla studie APOLLO-CD hodnotící účinnost a bezpečnost v rámci 12týdenní indukce u pacientů se střední až těžkou aktivní CD. Byl podán 55 pacientům, z nichž ve 12. týdnu dosáhlo endoskopické odpovědi 26 %. Nejčastější nežádoucí účinky byly covid-19, infekce močových cest, relaps CD, anemie, nazofaryngitida a únava [6,7]. Od června 2024 probíhá studie fáze III s názvem ARES-CD pro Crohnovu chorobu a studie ATLAS-UC pro ulcerózní kolitidu, jejichž kompletní výsledky budou známy v roce 2029.
Modulace mikroRNA-124
MikroRNA patří do skupiny nekódujících RNA molekul, které jsou zodpovědné za regulaci řady buněčných procesů včetně modulace transkripce a translace. Hlavní funkcí mikroRNA je blokáda translace vazbou na mRNA [8]. Molekula miR-124 reguluje expresi signální dráhy STAT3, která je zodpovědná za aktivaci makrofágů a zvýšení exprese prozánětlivých cytokinů, jako jsou TNFα nebo IFNγ. Tím přispívá k rozvoji střevního zánětu [9]. Ve střevní tkáni pacientů s IBD dochází ke zvýšení exprese STAT3 [10]. U petrických pacientů bylo zjištěno, že při aktivní UC dochází k downregulaci miR-124 a recipročně k nadměrné aktivaci STAT3 [11]. Upregulace miR- -124 vede ke snížení exprese STAT3, a tím k útlumu aktivace makrofágů ve střevě a k poklesu produkce proinflamatorních cytokinů [12,13]. V preklinických studiích na myších modelech IBD došlo po podání modulátoru miR-124 ke snížení produkce proinflamatorních cytokinů TNFα, IL-17 a IL-6. Byl také zaznamenán vzestup IL-22, který hraje významnou roli v hojení tkání a tlumení zánětu [14].
Obefazimod působí jako imunomodulátor prostřednictvím selektivní upregulace mikroRNA-124. Snižuje hladinu IL-17A a redukuje populaci Th17 buněk [14]. Pro UC indukční studie fáze III (ABTECT-1 a ABTECT-2) splnily primární cíle klinické remise v 8. týdnu při dávce 50 mg, přičemž prokázaly významně vyšší dosažení remise ve srovnání s placebem (celkem o 16,4 %) a dobrou snášenlivost. Pro Crohnovu chorobu probíhá studie fáze IIb ENHANCE-CD, jejíž výsledky budou dostupné v roce 2028. K výhodám obefazimodu patří perorální léková forma a jednoduché dávkování 1× denně. Nepůsobí přímo imunosupresivně, může být účinný i u pacientů po selhání biologické léčby a má příznivý bezpečnostní profil. Nejčastějšími pozorovanými nežádoucími účinky byly bolest hlavy a nauzea, které byly pravděpodobně závislé na dávce [13].
Stimulace receptoru NLRX1
Nucleotid oligomerization domain (NOD) like receptor X1 (NLRX1) je receptor nacházející se na vnější mitochondriální membráně, který potlačuje zánět a interferonovou odpověď na patogeny a inhibuje aktivitu NF-κB [15]. Kontroluje proliferaci a diferenciaci CD4+ T-lymfocytů. Při ztrátě jeho funkce dochází ke zvýšené diferenciaci T-lymfocytů do prozánětlivých fenotypů Th1 a Th17. Tyto buňky také častěji preferují anaerobní metabolizmus glukózy a jsou méně citlivé k regulaci prostřednictvím imunitních checkpointů. U myší s mutací NLRX1 byla pozorována vyšší aktivita zánětu a slizniční fibrózy ve střevech, vyšší hodnoty prozánětlivých cytokinů (IFNγ, TNFα, a IL-17) a Th1 a Th17 buněk oproti jejich zdravým protějškům [16,17].
Účinná látka NX-13 je perorální cílený agonista NLRX1 určený pro léčbu UC. Je podávána perorálně s minimální systémovou absorpcí, působí zejména ve střevní sliznici. Inhibuje diferenciaci CD4+ T-lymfocytů do Th1 a Th17 podtypů, což vede ke snížené produkci TNFα a INFγ. Má za sebou úspěšné studie fáze Ia/b, ve kterých byla prokázána bezpečnost a dobrá snášenlivost, takže byla zahájena II. fáze klinického hodnocení u středně těžké až těžké UC [18].
Inhibice receptor-interagujících proteinkináz
Receptor-interacting protein kinases 1–7 (RIPK 1–7) jsou skupina serin/threoninových a TKL (tyrosine kinase-like) kináz regulujících řadu buněčných procesů. RIPK1 má duální účinek, díky němuž za homestatických podmínek spouští proinflamatorní dráhu NF-κB a za podmínek patologických indukuje apoptózu prostřednictvím aktivace kaspázy-8. V případě, že jsou kaspázy inhibované a nedojde k aktivaci NF-κB, dochází prostřednictvím aktivovaných RIPK1 a RIPK3 k indukci nekroptózy [19]. Aktivace NF-κB prostřednictvím RIPK1 chrání intestinální epitelie a pomáhá udržovat střevní bariéru. Dysregulace RIPK drah ve střevních epiteliích vede k nadměrnému odumírání těchto buněk, uvolnění DAMPs (damage-associated molecular patterns) a amplifikaci zánětu [20]. Exprese RIPK3 koreluje se závažností a aktivitou CD [21].
Látka GSK2982772 je cílený RIPK1 inhibitor, který má bránit apoptóze střevního epitelu a zánětlivé infiltraci sliznice a chránit bariérovou funkci střeva prostřednictvím inhibice drah TNFα a NF-κB. Ve studii fáze IIa u pacientů s UC byla prokázána dobrá snášenlivost bez závažných nežádoucích účinků, s vyššími koncentracemi ve střevní tkáni než v séru. Účinnost na aktivitu onemocnění však prokázána nebyla [22].
Blokáda receptoru pro interleukin 36
Interleukin 36 je spolu s IL-33 a IL-38 členem cytokinové rodiny IL-1. Všechny tyto cytokiny hrají roli v patogenezi IBD. Vazbou IL-36 na svůj receptor IL-36R dochází ke stimulaci drah NF-κB a MAPK (mitogen-activated protein kinase) a k následné produkci IL-6 a TNFα. IL-36 je vylučován keratinocyty, T- a B-lymfocyty, monocyty, makrofágy, epiteliemi a fibroblasty. Sekrece monocytů a makrofágů nastává po stimulaci ligandy Toll-like receptorů fibroblastů kombinací IL-1β a TNFα. U epitelií a T buněk je produkce IL-36 spuštěna epidermálními růstovými faktory (EGFs) a růstovými faktory fibroblastů (FGFs) [23]. Ve sliznici pacientů s CD a UC byla popsána zvýšená exprese IL-36 [24].
Spesolimab je humanizovaná monoklonální protilátka proti receptoru pro IL-36, v současnosti schválená pro léčbu generalizované pustulózní psoriázy. Je ve fázi II/III vývoje pro léčbu IBD. Klinické studie pro UC ukázaly, že je sice dobře snášen, ale cíle účinnosti nebyly vždy splněny, takže jeho vývoj pokračuje s cílem nalezení optimální dávky a potvrzení účinnosti u pacientů, kteří nereagovali na jiné typy léčby [25].
Inhibice transsignální dráhy interleukinu 6
Interleukin 6 je cytokin, k jehož expresi dochází v reakci na infekci a poškození tkání. Jeho dysregulovaná produkce hraje roli v patogenezi chronického zánětu a autoimunitních chorob [26]. IL-6 má tři cesty signalizace, jimiž jsou klasická, trans a cluster signalizace. Každá z těchto cest má v organizmu odlišnou funkci. Klasická cesta signalizace spouští pochody regenerační a ochranné, jako je inhibice apoptózy epitelií a stimulace jejich proliferace, aktivace reakce akutní fáze v játrech a ochrana proti bakteriálním infekcím. Transsignalizace na druhou stranu spouští děje proinflamatorní, jako je inhibice apoptózy T-lymfocytů, stimulace migrace monocytů, stimulace endotelií a buněk hladké svaloviny a inhibice diferenciace Treg buněk. Cluster signalizace hraje roli v aktivaci patologických Th17 buněk.
Cílená inhibice pouze transsignalizace slibuje nižší míru imunosuprese a zachování regeneračních procesů probíhajících pod vlivem klasické cesty signalizace IL-6 [27]. Transsignalizace probíhá vazbou komplexu IL-6 a volného receptoru pro IL-6 (sIL-6R) na membránově vázaný glykoprotein gp130, který je klíčovou transmembránovou signální podjednotkou řady receptorů. Spouští tak reakce na buňkách, které nemají na svém povrchu receptor pro IL-6 (IL-6R). Klasická signalizace spočívá ve vazbě IL-6 na membránově vázaný IL-6R, který je exprimován spolu s gp130 [28].
Olamkicept je fúzní protein složený ze dvou extracelulárních domén gp130 a jednoho Fc fragmentu lidského IgG1. Váže pouze komplexy tvořené IL-6 a sIL-6R, čímž zabraňuje jejich vazbě na buňky s membránově vázaným gp130, a blokuje tak pouze transsignalizaci. Má dokončeny klinické studie fáze II se slibnými výsledky u UC i CD v dávce 600 mg i.v. podávané à 2 týdny [29].
Inhibice fosfodiesterázy 4
Fosfodiesteráza 4 (PDE4) je skupina enzymů degradující cyklický adenosinmonofosfát (cAMP). Důsledkem je zvýšení produkce pro-inflamatorních cytokinů (TNFα, IL-23) a snížení protizánětlivých cytokinů (IL-10). Inhibice PDE4 zvyšuje koncentraci cAMP a následně zvyšuje expresi protizánětlivých cytokinů. V současnosti jsou u nás schváleny pro léčbu psoriatické artritidy, psoriázy a Behçetovy nemoci. Proběhla klinická studie fáze II s apremilastem u pacientů s UC, jejíž výsledky však byly neprůkazné [30]. Novější inhibitory cílené na GIT jsou zatím v preklinických a časných klinických fázích vývoje.
Stimulace receptoru pro melanokortin 1 (MC1R)
Melanokortiny jsou skupina čtyř peptidových hormonů: ACTH, α-, β- a γ-melanocyty stimulující hormony (MSH). Je známo pět podtypů melanokortinových receptorů (MC1-5R). Jsou to membránové receptory spřažené s G-proteinem. MC1R se nachází zejména v melanocytech a leukocytech. Přirozeným agonistou MC1R je α-MSH. Jeho vazba na tento receptor vede ke zvýšení produkce cAMP, aktivaci protein kinázy A a C a ve výsledku k aktivaci MAPK (mitogen-activated protein kinase) a JAK-STAT drah. Důsledkem je útlum exprese prozánětlivých cytokinů (TNFα, IL-1, IL-6) prostřednictvím inhibice NF-κB signální dráhy a upregulace protizánětlivého IL-10 [31,32].
Látka PL8177 je selektivním agonistou MC1R a v současnosti s ní probíhá klinická studie fáze II. Testuje se jeho podávání ve formě s řízeným uvolňováním v tlustém střevě s minimální systémovou absorpcí, a tedy i nežádoucími účinky [32].
Inhibice interleukinů 1α a 1β
Interleukin 1 je skupina jedenácti cytokinů hrajících zásadní roli v modulaci zánětu a dalších procesů zprostředkovaných vrozenou imunitou. Jsou produkovány širokou škálou imunitních buněk, epitelií i keratinocytů v reakci na PAMPs a DAMPS. Vazba na receptor pro IL-1 vede k aktivaci NF-κB a MAPK signálních drah. IL-1α i IL-1β jsou exprimovány ve střevních epiteliích. IL-1α se nachází běžně v epiteliích pacientů s IBD a je uvolňován z nekrotického střevního epitelu a stimuluje fibroblasty k produkci IL-6 a IL-8 (33). IL-1β indukuje expresi adhezivních molekul na endoteliích, a tím podporuje maturaci dendritických buněk. Funguje také jako endogenní pyrogen a facilituje diferenciaci Th17 buněk. Jeho zvýšené hladiny u pacientů s IBD jsou asociovány s chronickým zánětem a těžším průběhem choroby [34].
V současnosti probíhají klinické studie fáze II u pacientů s UC a CD s účinnou látkou lutikizumab. Jedná se o duální monoklonální protilátku proti IL-1α i β. Výsledky studií zatím nebyly publikovány, avšak podle sdělení výrobce v monoterapii UC sice prokázal lepší výsledky než kontrolní adalimumab, avšak rozdíl nebyl natolik významný, aby výzkum v této indikaci pokračoval. Nyní je testován v kombinační léčbě CD.
Blokáda receptoru pro interleukin 31 a onkostatin M
Onkostatin M (OSM) patří do skupiny IL-6 cytokinů. Je produkován hematopoetickými buňkami a váže se zejména na povrchu fibroblastů a epitelií na svůj receptor OSMR. OSMR je heterodimer skládající se z podjednotky OSMRβ a gp130. Tato vazba vede ke spuštění drah STAT3, MAPK a PI3K. Má pro- i protizánětlivé účinky a hraje roli v řadě patologických procesů jako revmatoidní artritida, psoriáza nebo infekce. Jeho zvýšená exprese byla nalezena v bioptických vzorcích IBD pacientů a uvažuje se o něm jako o možném prediktoru primární rezistence k léčbě anti-TNF protilátkami [35,36]. Ve střevní tkáni pacientů s aktivním IBD byla nalezena zvýšená exprese OSM a jeho receptoru. Pacienti s nízkou hladinou OSM v plazmě před zahájením léčby měli vyšší šanci na dosažení klinické remise do 1 roku po anti-TNF terapii [37]. IL-31 patří také do skupiny IL-6 cytokinů. S onkostatinem M sdílí receptorovou podjednotku OSMRβ. Je produkován CD4+ pomocnými T-lymfocyty, žírnými buňkami, monocyty/makrofágy a dendritickými buňkami. Hraje roli v patogenezi alergických onemocnění a v rozvoji pruritu [38].
Vixarelimab je monoklonální protilátka proti β-podjednotce OSM receptoru, která blokuje vazbu OSM a IL-31. Klinická studie fáze II u pacientů se středně těžkou až těžkou UC však byla zastavena pro nepravděpodobnost dosažení primárních cílů a výzkum pokračuje pouze v indikacích s nadějnějšími dosavadními výsledky, jako je prurigo nodularis.
Inhibice tyrosinkinázy SYK
Spleen tyrosin kinase (SYK) je nereceptorová tyrosinkináza nacházející se v cytoplazmě hematopoetických (T- a B-lymfocyty, dendritické buňky, makrofágy, žírné buňky) i jiných buněk (střevní epitelie, endotelie, fibroblasty, hepatocyty) [39]. U B-lymfocytů zajišťuje přenos signálů z povrchových receptorů a je nezbytná pro jejich diferenciaci a vyzrávání. Za normálních okolností je SYK neaktivní. Po stimulaci receptoru (BCR, TCR) dochází k její aktivaci a následně k fosforylaci řady adaptorových proteinů v cytoplazmě lymfocytu, které pak spouští další signální kaskády, jako je NF-κB, MAPK, NFAT. Myši s inaktivovaným genem pro SYK postrádají zralé B-lymfocyty. Naopak nadměrná aktivace SYK vede k hypogamaglobulinemii a zvýšené produkci proinflamatorních cytokinů. SYK tak hraje důležitou roli v rozpoznávání vlastního od cizího, což je jeden z patogenetických mechanizmů zodpovědných za rozvoj IBD [40].
Lanraplenib je malá molekula cílená k inhibici SYK. Stejně jako u jiných tyrosinkinázových inhibitorů je jeho výhodou možnost perorálního podávání jednou denně. V současnosti probíhá klinická studie fáze II v indikaci UC.
Stimulace PD-1 checkpointu
Imunitní checkpointy neboli kontrolní body jsou skupina inhibičních nebo stimulačních molekul exprimovaných na povrchu imunitních buněk, APCs a nádorových buněk, jejichž funkcí je modulace aktivace T-lymfocytů. Programmed cell death 1 protein (PD-1) je transmembránový protein nacházející se na povrchu aktivovaných T-lymfocytů, B-lymfocytů, NK buněk, makrofágů, dendritických buněk, monocytů a myeloidních buněk [41]. Jeho imunosupresivní funkce zahrnují inhibici proliferace, aktivace, produkce cytokinů a změnu metabolizmu T buněk vedoucí až ke smrti aktivovaných T-lymfocytů [42]. Deficit PD-1 u myší vede k rozvoji autoimunitních chorob [43]. K imunosupresi dochází po vazbě jednoho ze dvou hlavních ligandů PD-L1 nebo PD-L2 exprimovaných na povrchu buněk imunitního systému i řady dalších buněk (nádorové buňky, epitelie, endotelie). PD-1 checkpoint pomáhá navozovat a udržovat toleranci imunitního systému vůči vlastním antigenům [41].
Pacienti s preexistujícím IBD léčení PD-1 inhibitory pro maligní nádory mají vyšší riziko rozvoje gastrointestinálních nežádoucích účinků, jako je průjem, kolitida nebo i perforace střeva [44]. Navíc bylo zjištěno, že u pacientů s IBD byly v krvi a v zánětem postižených sliznicích nalezeny T buňky exprimující PD-1, které jsou možným pozitivním prediktorem odpovědi na léčbu vedolizumabem [45].
Rosnilimab je monoklonální protilátka s agonistickým účinkem na PD-1. Redukuje proliferaci a sekreci cytokinů u T buněk s vysokou expresí PD-1 u zdravých dobrovolníků [46]. Koncem roku 2025 však bylo oznámeno, že studie fáze II u pacientů UC neprokázala klinickou účinnost po 12 týdnech léčby, a proto byla zastavena. Rosnilimab v ní sice skutečně redukoval počty zánětlivých buněk a cytokinů, nicméně to nebylo dostačující k dosažení klinické remise. V indikaci revmatoidní artritida však naopak klinická účinnost prokázána byla, takže studium tohoto terapeutického cíle bude pokračovat.
Inhibice reninu
Systém renin-angiotenzin-aldosteron (RAAS) reguluje krevní tlak a homeostázu tekutin v organizmu. Za normálních okolností také reguluje rovnováhu tekutin a elektrolytů, absorpci živin a exkreci bikarbonátů ve střevě. Bylo prokázáno že v zánětem postiženém střevě dochází ke změnám funkce a hladin látek RAAS. Patologická aktivace RAAS ve střevě vede ke zvýšení produkce angiotenzinu II, který následně působí prozánětlivě skrze aktivaci dráhy NF-κB, proapoptoticky a profibroticky skrze TGF-β1. Vede také k indukci oxidativního stresu a aktivaci neutrofilů a makrofágů [47]. U myších modelů IBD byla prokázána upregulace reninu, ACE, AT1R a angiotenzinu II ve střevě. Nadměrná exprese reninu vedla u těchto myší ke zhoršení kolitidy a zvýšené apoptóze epitelií [48]. Role RAAS u IBD je poměrně dobře prozkoumána na zvířecích modelech.
Protizánětlivé a antifibrotické účinky po podání inhibitorů RAAS, jako jsou ACE inhibitory, sartany a inhibitory reninu, byly prokázány v experimentech na zvířatech [47]. U člověka se zatím můžeme opřít o retrospektivní studie srovnávající pacienty s IBD užívající a neužívající ACE inhibitory nebo sartany. Bylo zjištěno, že pacienti užívající ACEi nebo ARB měli méně hospitalizací, operací a nižší potřebu užívání glukokortikoidů [49].
V indikaci IBD je testován inhibitorem reninu sitokiren (SPH3127), zatím ve fázi II klinického hodnocení v indikaci lehké až středně těžké UC. V preklinických testech prokázal slibné výsledky, jako je snížení produkce proinflamatorních cytokinů (TNFα, IL-1β, IL-6, IFNγ, IL-17) a zvýšení produkce protizánětlivého IL-10 ve střevě. Tlumí také aktivaci slizničních Th1 a Th17 buněk [50].
Blokáda receptorů CXCR
CXCR1 a 2 jsou chemokinové receptory exprimované na povrchu leukocytů (neutrofily, makrofágy, žírné buňky), endotelií a nádorových buněk. Na tyto receptory se váže sedm ligandů CXCL1-3 a CXCL5-8. Nejlépe prozkoumaným ligandem těchto receptorů je CXCL8 také známý jako IL-8. CXCL8 je produkován monocyty, makrofágy, fibroblasty, hepatocyty, epiteliemi a endoteliemi. Jeho sekrece je stimulována TNFα nebo IL-1β. Po vazbě na CXCR1/2 je zodpovědný za aktivaci a chemotaxi neutrofilů do místa zánětu a zvyšuje produkci kyslíkových radikálů [51]. Další funkcí neutrofilů je tvorba NETů (neutrophil extracellular traps). Jedná se o extracelulární sítě, jejichž funkcí je zadržení a eliminace potenciálních patogenů. U pacientů s UC byly nalezeny vyšší hladiny NETů v periferní krvi oproti zdravým jedincům. V biopsiích střeva pacientů s UC byla nalezena vyšší exprese CXCR1/2 i jejich ligandu CXCL8 [52].
Monoklonální protilátka eltrekibart (LY3041658) neutralizuje všech sedm ligandů CXCR1/2 [53]. Blokuje tak migraci neutrofilů, aniž by blokovala jejich funkce (aktivace, oxidativní vzplanutí, fagocytóza). V současnosti probíhá klinická studie fáze II u UC v kombinaci s mirikizumabem.
Stimulace receptoru PSGL-1
PSGL-1 (P-selectin glykoprotein ligand-1) je transmembránový receptor exprimovaný na povrchu všech leukocytů. Jeho ligandy jsou P-, E- a L-selektiny. Vazba PSGL-1 (na povrchu leukocytu) na P-selektin (na endotelu) vede k adhezi leukocytu na aktivovaný endotel a jeho přesunu do místa zánětu [54]. PSGL-1 také funguje jako imunitní checkpoint tlumící aktivitu T-lymfocytů a reguluje imunitní odpovědi tak, aby nedocházelo k přehnané reaktivitě. Snižuje reaktivitu na TCR signalizaci, pomocí které T-lymfocyt rozpoznává antigenní peptidy.
Agonizmus na PSG-1 je proto považován za možný terapeutický cíl u autoimunitních nebo zánětlivých onemocnění s nadměrnou aktivací T-buněk, zatímco antagonizmus může být využit v léčbě nádorových onemocnění [55]. V preklinických studiích prokázaly agonistické protilátky na PSG-1 schopnost navodit apoptózu aktivovaných T-buněk u modelů GvHD a DM 1. typu, aniž by byly ovlivněny ostatní T buňky [56].
V současné době probíhá studie fáze II u UC s přípravkem ALTB-268. Jedná se o podkožně podávanou tetravalentní monoklonální protilátku, která je agonistou PSGL-1 [57].
Inhibice NLRP3 inflamazomu
Cytoplazmatické tzv. NOD-like receptory fungují jako PRRs (pattern recognition receptors). Pokud dojde k jejich aktivaci prostřednictvím DAMPs (damage-associated molecular patterns) nebo PAMPs (patogen-associated molecular patterns), jsou schopny se poskládat do makromolekulárních komplexů zvaných inflamazomy. Hlavní funkcí inflamazomů je aktivace specifických kaspáz a následně spuštění zánětlivé odpovědi. NOD-like receptor protein 3 (NLRP3) vytváří NLRP3 inflamazom, který vede k aktivaci kaspázy-1 a k produkci prozánětlivých cytokinů IL-1β a IL-18 [58]. Vysoká koncentrace IL-1β ve střevní tkáni pacientů s IBD koreluje s aktivitou choroby. IL-1β hraje roli v patogenezi IBD prostřednictvím zvyšování permeability epitelu a stimulací produkce dalších proinflamatorních cytokinů (IL-6). Inhibice NLRP3 vede ke snížení koncentrace IL-1β a IL-18 [59]. Ve střevní sliznici pacientů s CD byla nalezena upregulace a zvýšená aktivita NLRP3 inflamazomu, což z něj činí možný terapeutický cíl [60].
Usnoflast (ZYIL1) je perorální inhibitor NLRP3 inflamazomu. Proběhla klinická studie fáze IIa u pacientů s lehkou až středně těžkou UC, kterým byl usnoflast podáván per os dvakrát denně. Výsledky zatím nebyly publikovány [61]. Tato látka je testována i v terapii amyotrofické laterální sklerózy jako orphan drug.
Inhibice histonové deacetylázy
Acetylace histonů je jedním z mechanizmů posttranslační modifikace histonů. Za odstranění acetylové skupiny, a tím za kondenzaci chromatinu a útlum genové exprese je zodpovědná skupina enzymů zvaná histonové deacetylázy (HDAC). U člověka je identifikováno 18 izoforem HDAC, které jsou rozděleny do pěti tříd. HDAC regulují širokou škálu buněčných procesů, jako je apoptóza, proliferace a diferenciace. Jsou také zapojeny do široké škály imunitních procesů včetně syntézy a sekrece cytokinů [62]. Studie ukazují, že inhibice HDAC zasahuje různými způsoby do patogeneze IBD. Indukuje diferenciaci Treg buněk, inhibuje diferenciaci Th17 buněk, monocytů a makrofágů a signální dráhu NF-κB [63].
Vorinostat je inhibitorem HDAC třídy I a II. Je v současnosti testován v dávce 100 mg per os dvakrát denně v léčbě středně těžké až těžké CD v kombinaci s ustekinumabem.
Substituce interleukinu 2
Interleukin 2 je cytokin produkovaný CD4+ T-lymfocyty. Váže se na IL-2 receptor (IL-2R) na povrchu Treg buněk, aktivovaných T-lymfocytů, NK buněk a naivních a paměťových T buněk. Jeho funkce spočívá v udržení imunitní tolerance prostřednictvím stimulace růstu a přežívání Treg buněk. Ty jsou nezbytné pro udržení tolerance vůči vlastním antigenům prostřednictvím kontroly aktivace, diferenciace a expanze autoreaktivních T buněk. Nedostatek IL-2 tak vede k narušení homeostázy Treg buněk, a tím ke snížení imunitní tolerance [64]. Vysoké dávky analog IL-2 mají protinádorové účinky, ale jsou zatíženy závažnými nežádoucími účinky, jako je syndrom zvýšené propustnosti kapilár [65]. Nízké dávky byly testovány u pacientů se širokou škálou autoimunitních onemocnění včetně IBD a prokázaly bezpečnost a schopnost expanze a aktivace populace Treg buněk. U pacientu s UC došlo ke snížení hodnoty Mayo skóre [66].
Aldesleukin je analog IL-2 používaný původně u maligního melanomu a karcinomu ledviny. U pacientů se středně těžkou až těžkou UC již proběhla fáze I/II klinického testování subkutánního podávání nízkých dávek s dobrou tolerancí a u pacientů s CD stále probíhá [67].
Blokáda receptoru typu I pro transformující růstový faktor β
Transforming growth factor-β (TGF-β) je cytokin exprimovaný v téměř všech buňkách v organizmu. Jeho nejdůležitějšími receptory jsou receptor typu I (TGFBR1), také známý jako ALK5, a receptor typu II (TGFBR2). TGF-β je za fyziologických podmínek důležitý pro embryonální vývoj, hojení tkání a homeostázu tkání i imunity. Jeho dysfunkce hraje roli v řadě patologických procesů, jako je vznik nádorů, zánětlivých onemocnění, infekčních onemocnění a fibrózních procesů [68]. Za patologických podmínek produkuje řada buněk ve větším množství TGF-β, který následně podporuje akumulaci extracelulární matrix a rozvoj fibrózy [69]. Inhibitory ALK5 představují jednu z možností farmakologického zásahu do procesu fibrózy, který představuje zdroj komplikací zejména u CD.
Ontunisertib (AGMB-129) je malá molekula účinkující jako ALK5 inhibitor. Je testován pro léčbu fibrostenotické CD v denní dávce 400 mg per os s povzbudivými předběžnými výsledky [70].
Blokáda receptoru CCR9
C-C chemokine receptor 9 (CCR9) je chemokinový receptor nacházející se na povrchu dendritických buněk, neutrofilů, lymfocytů, monocytů, makrofágů a endotelií, zejména však na povrchu nezralých T-lymfocytů. Jeho ligandem je C-C chemokine ligand 25 (CCL25) exprimovaný v thymu a intestinálních epiteliích [71]. Aktivované T-lymfocyty exprimující na svém povrchu CCR9 a α4β7 integrin jsou po vazbě na CCL25 na střevním epitelu preferenčně přesouvány do střevní tkáně, kde tvoří zánětlivý infiltrát. Většina T buněk exprimující CCR9 jsou producenti IFNγ, menší část z nich pak produkuje IL-17A, IL-10 a IL-4. U některých pacientů s CD bylo v periferní krvi nalezeno zvýšené množství CCR9+ T buněk [72]. U zvířecích modelů IBD měla inhibice vazby CCR9/CCL25 za následek zmírnění intenzity zánětu. V minulosti testované inhibitory CCR9 však svou účinnost ve studiích neprokázaly, pravděpodobně z důvodu nepříznivého farmakokinetického profilu [73].
AZD7798 je monoklonální protilátka proti CCR9. Prokázala schopnost snížit množství CCR9+ buněk ve střevě. Probíhá s ní klinická studie fáze II u pacientů s CD [74].
Závěr
Z výše uvedeného přehledu, z důvodů rozsahu rukopisu stručného a zcela jistě nekompletního je zřejmé, jak velké úsilí a zdroje jsou výzkumu farmakoterapie IBD věnovány. Podle našeho názoru je však zavedení převratně účinné farmakoterapie v nejbližší budoucnosti velmi nepravděpodobné. Mezi nově testovanými cíli zcela zjevně absentuje takový, který by byl zcela specifický pro IBD. Většinou se jedná o nově testované cíle v kaskádě imunitní reakce, které z principu mohou přinést vyšší bezpečnost, ale jen stěží zásadně vyšší účinnost z hlediska klinického průběhu onemocnění. To potvrzuje i fakt, že většina zmíněných účinných látek je testována nebo již zavedena v terapii jiných autoimunit. Navíc průměrná úspěšnost přechodu testovaného léku z II. do III. fáze klinického hodnocení se v současnosti pohybuje pouze kolem 30 %. Vývoj některých látek zmíněných v textu byl již zastaven, což ovšem zcela nevylučuje úspěch dalšího výzkumu v působení na původní farmakodynamický cíl. Nicméně se zdá, že dokud nebude nalezen cíl zcela specifický pro IBD (a pravděpodobně i jiný pro CD a jiný pro UC), nedojde k zavedení farmakoterapie se skutečně kurativním účinkem. To by ovšem v podstatě znamenalo odhalení příčiny obou nemocí. Nicméně vzhledem k závažnosti těchto onemocnění a jejich celosvětovému nárůstu nelze pochybovat o tom, že i malý pokrok ve farmakoterapii může znamenat pro nemocné značný přínos.
ORCID autora
K. Urbánek 0000-0002-3461-1649.
Doručeno/Submitted: 8. 3. 2026
Přijato/Accepted: 15. 3. 2026
Korespondenční autorka
MUDr. Petra Hlaušková
II. interní klinika – gastroenterologická a geriatrická
LF UP a FN Olomouc
Zdravotníků 248/7
779 00 Olomouc
petra.hlauskova@fnol.cz
Literatura
1. Ng SC, Shi HY, Hamidi N et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet 2017; 390(10114): 2769–2778. doi: 10.1016/S0140-6736(17)32448-0.
2. Petronio L, Dal Buono A, Gabbiadini R et al. Drug development in inflammatory bowel diseases: what is next? Pharmaceuticals (Basel) 2025; 18(2): 190. doi: 10.3390/ph18020190.
3. Valatas V, Kolios G, Bamias G. TL1A (TNFSF15) and DR3 (TNFRSF25): a co-stimulatory system of cytokines with diverse functions in gut mucosal immunity. Front Immunol 2019; 10: 583. doi: 10.3389/fimmu.2019.00583.
4. Varricchi G, Poto R, Criscuolo G et al. TL1A, a novel alarmin in airway, intestinal, and autoimmune disorders. J Allergy Clin Immunol 2025; 155(5): 1420–1434. doi: 10.1016/ j.jaci.2025.02.018.
5. Solitano V, Jairath V, Ungaro F et al. TL1A inhibition for inflammatory bowel disease treatment: from inflammation to fibrosis. Med 2024; 5(5): 386–400. doi: 10.1016/j.medj.2024.03.010.
6. Feagan BG, Sands BE, Siegel CA et al. Safety and efficacy of the anti-TL1A monoclonal antibody tulisokibart for Crohn’s disease: a phase 2a induction trial. Lancet Gastroenterol Hepatol 2025; 10(8): 715–725. doi: 10.1016/S2468-1253(25)00071-8.
7. Sands BE, Feagan BG, Peyrin-Biroulet L et al. Phase 2 trial of anti-TL1A monoclonal antibody tulisokibart for ulcerative colitis. N Engl J Med 2024; 391(12): 1119–1129. doi: 10.1056/NEJMoa2314076.
8. Suri K, Bubier JA, Wiles MV et al. Role of microRNA in inflammatory bowel disease: clinical evidence and the development of preclinical animal models. Cells 2021; 10(9): 2204. doi: 10.3390/cells10092204.
9. Tazi J, Begon-Pescia C, Campos N et al. Specific and selective induction of miR-124 in immune cells by the quinoline ABX464: a transformative therapy for inflammatory diseases. Drug Discov Today 2021; 26(4): 1030–1039. doi: 10.1016/j.drudis.2020.12.019.
10. Sugimoto K. Role of STAT3 in inflammatory bowel disease. World J Gastroenterol 2008; 14(33): 5110–5114. doi: 10.3748/wjg.14.5110.
11. Koukos G, Polytarchou C, Kaplan JL et al. MicroRNA-124 regulates STAT3 expression and is down-regulated in colon tissues of petric patients with ulcerative colitis. Gastroenterology 2013; 145(4): 842–852.e2. doi: 10.1053/j.gastro.2013.07.001.
12. Xiao YT, Wang J, Lu W et al. Downregulated expression of microRNA-124 in petric intestinal failure patients modulates macrophages activation by inhibiting STAT3 and AChE. Cell Death Dis 2016; 7(12): e2521. doi: 10.1038/cddis.2016.426.
13. Vermeire S, Solitano V, Peyrin-Biroulet L et al. Obefazimod: a first-in-class drug for the treatment of ulcerative colitis. J Crohns Colitis 2023; 17(10): 1689–1697. doi: 10.1093/ecco-jcc/jjad067.
14. Apolit C, Campos N, Vautrin A et al. ABX464 (obefazimod) upregulates miR-124 to reduce proinflammatory markers in inflammatory bowel diseases. Clin Transl Gastroenterol 2023; 14(4): e00560. doi: 10.14309/ctg.0000000000000560.
15. Nagai-Singer MA, Morrison HA, Allen IC. NLRX1 is a multifaceted and enigmatic regulator of immune system function. Front Immunol 2019; 10: 2419. doi: 10.3389/fimmu.2019.02419.
16. Leber A, Hontecillas R, Zoccoli-Rodriguez V et al. Activation of NLRX1 by NX-13 alleviates inflammatory bowel disease through immunometabolic mechanisms in CD4+ T cells. J Immunol 2019; 203(12): 3407–3415. doi: 10.4049/jimmunol.1900364.
17. Leber A, Hontecillas R, Tubau-Juni N et al. NLRX1 regulates effector and metabolic functions of CD4+ T cells. J Immunol 2017; 198(6): 2260–2268. doi: 10.4049/jimmunol.1601547.
18. Verstockt B, Vermeire S, Peyrin-Biroulet L et al. The safety, tolerability, pharmacokinetics, and clinical efficacy of the NLRX1 agonist NX-13 in active ulcerative colitis: results of a phase 1b study. J Crohns Colitis 2024; 18(5): 762–772. doi: 10.1093/ecco-jcc/jjad192.
19. Garcia-Carbonell R, Yao SJ, Das S et al. Dysregulation of intestinal epithelial cell RIPK pathways promotes chronic inflammation in the IBD gut. Front Immunol 2019; 10: 1094. doi: 10.3389/fimmu.2019.01094.
20. Degterev A, Ofengeim D, Yuan J. Targeting RIPK1 for the treatment of human diseases. Proc Natl Acad Sci U S A 2019; 116(20): 9714–9722. doi: 10.1073/pnas.1901179116.
21. Panayotova E, Atanassova A. P043 RIPK3 – a new marker for assessment of activity in Crohn’s disease. J Crohns Colitis 2024; 18(Suppl 1): i301. doi: 10.1093/ecco-jcc/jjad212.0173.
22. Weisel K, Scott N, Berger S et al. A randomised, placebo-controlled study of RIPK1 inhibitor GSK2982772 in patients with active ulcerative colitis. BMJ Open Gastroenterol 2021; 8(1): e000680. doi: 10.1136/bmjgast-2021-000680.
23. Andoh A, Nishida A. Pro- and anti-inflammatory roles of interleukin (IL) -33, IL-36, and IL-38 in inflammatory bowel disease. J Gastroenterol 2023; 58(2): 69–78. doi: 10.1007/s00535-022-01936-x.
24. Nishida A, Hidaka K, Kanda T et al. Increased expression of interleukin-36, a member of the interleukin-1 cytokine family, in inflammatory bowel disease. Inflamm Bowel Dis 2016; 22(2): 303–314. doi: 10.1097/MIB.0000000000000654.
25. Ferrante M, Irving PM, Selinger CP et al. Safety and tolerability of spesolimab in patients with ulcerative colitis. Expert Opin Drug Saf 2023; 222: 141–152. doi: 10.1080/14740338.2022.2103536.
26. Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol 2014; 6(10): a016295. doi: 10.1101/cshperspect.a016295.
27. Rose-John S, Jenkins BJ, Garbers C et al. Targeting IL-6 trans-signalling: past, present and future prospects. Nat Rev Immunol 2023; 23(10): 666–681. doi: 10.1038/s41577-023-00856-y.
28. Jostock T, Müllberg J, Ozbek S et al. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur J Biochem 2001; 268(1): 160–167. doi: 10.1046/j.1432-1327.2001.01867.x.
29. Schreiber S, Aden K, Bernardes JP et al. Therapeutic interleukin-6 trans-signaling inhibition by olamkicept (sgp130Fc) in patients with active inflammatory bowel disease. Gastroenterology 2021; 160(7): 2354–2366. doi: 10.1053/j.gastro.2021.02.062.
30. Danese S, Neurath MF, Kopoń A et al. Effects of apremilast, an oral inhibitor of phosphodiesterase 4, in a randomized trial of patients with active ulcerative colitis. Clin Gastroenterol Hepatol 2020; 18(11): 2526–2534.e9. doi: 10.1016/j.cgh.2019.12.032.
31. Mun Y, Kim W, Shin D. Melanocortin 1 receptor (MC1R): pharmacological and therapeutic aspects. Int J Mol Sci 2023; 24(15): 12152. doi: 10.3390/ijms241512152.
32. Dodd J, Jordan R, Makhlina M et al. A novel oral formulation of the melanocortin-1 receptor agonist PL8177 resolves inflammation in preclinical studies of inflammatory bowel disease and is gut restricted in rats, dogs, and humans. Front Immunol 2023; 14: 1083333. doi: 10.3389/fimmu.2023.1083333.
33. Scarpa M, Kessler S, Sadler T et al. The epithelial danger signal IL-1α is a potent activator of fibroblasts and reactivator of intestinal inflammation. Am J Pathol 2015; 185(6): 1624–1637. doi: 10.1016/j.ajpath.2015.02.018.
34. Aggeletopoulou I, Kalafateli M, Tsounis EP et al. Exploring the role of IL-1β in inflammatory bowel disease pathogenesis. Front Med (Lausanne) 2024; 11: 1307394. doi: 10.3389/fmed. 2024.1307394.
35. West NR, Hegazy AN, Owens BMJ et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat Med 2017; 23(5): 579–589. doi: 10.1038/nm.4307.
36. Kokkotis G, Filidou E, Tarapatzi G et al. Oncostatin M induces a pro-inflammatory phenotype in intestinal subepithelial myofibroblasts. Inflamm Bowel Dis 2024; 30(11): 2162–2173. doi: 10.1093/ibd/izae098.
37. Guo A, Ross C, Chande N et al. High oncostatin M predicts lack of clinical remission for patients with inflammatory bowel disease on tumor necrosis factor α antagonists. Sci Rep 2022; 12(1): 1185. doi: 10.1038/s41598-022-05208-9.
38. Hermanns HM. Oncostatin M and interleukin-31: cytokines, receptors, signal transduction and physiology. Cytokine Growth Factor Rev 2015; 26(5): 545–558. doi: 10.1016/ j.cytogfr.2015.07.006.
39. Yang Y, Wang L, Zeng Z et al. Gain-of-function variant in spleen tyrosine kinase regulates macrophage migration and functions to promote intestinal inflammation. J Inflamm Res 2024; 17: 8713–8726. doi: 10.2147/JIR.S 488901.
40. Aksentijevich I. The sickening consequences of too much SYK signaling. Nat Genet 2021; 53(4): 432–434. doi: 10.1038/s41588-021-00837-8.
41. Chen RY, Zhu Y, Shen YY et al. The role of PD-1 signaling in health and immune-related diseases. Front Immunol 2023; 14: 1163633. doi: 10.3389/fimmu.2023.1163633.
42. Ghosh C, Luong G, Sun Y. A snapshot of the PD-1/PD-L1 pathway. J Cancer 2021; 12(9): 2735–2746. doi: 10.7150/jca.57334.
43. Yoshida T, Jiang F, Honjo T et al. PD-1 deficiency reveals various tissue-specific autoimmunity by H-2b and dose-dependent requirement of H-2g7 for betes in NOD mice. Proc Natl Acad Sci U S A 2008; 105(9): 3533–3538. doi: 10.1073/pnas.0710951105.
44. Abu-Sbeih H, Faleck DM, Ricciuti B et al. Immune checkpoint inhibitor therapy in patients with preexisting inflammatory bowel disease. J Clin Oncol 2020; 38(6): 576–583. doi: 10.1200/JCO.19.01674.
45. Kim MK, Jo SI, Kim SY et al. PD-1-positive cells contribute to the gnosis of inflammatory bowel disease and can aid in predicting response to vedolizumab. Sci Rep 2023; 13(1): 21329. doi: 10.1038/s41598-023-48651-y.
46. Luu K, Dahl M, Hare E et al. DOP81 rosnilimab, a novel PD-1 agonist monoclonal antibody, reduces T cell proliferation, inflammatory cytokine secretion, and PD-1high expressing CD4 and CD8 T cells: results from a Phase 1 healthy volunteer clinical trial. J Crohns Colitis 2025; 18(Suppl 1): i225. doi: 10.1093/ecco-jcc/jjad212.0121.
47. Salmenkari H, Korpela R, Vapaatalo H. Renin-angiotensin system in intestinal inflammation-angiotensin inhibitors to treat inflammatory bowel diseases? Basic Clin Pharmacol Toxicol 2021; 129(3): 161–172. doi: 10.1111/bcpt.13624.
48. Shi Y, Liu T, He L et al. Activation of the renin-angiotensin system promotes colitis development. Sci Rep 2016; 6: 27552. doi: 10.1038/srep 27552.
49. Jacobs JD, Wagner T, Gulotta G et al. Impact of angiotensin II signaling blockade on clinical outcomes in patients with inflammatory bowel disease. Dig Dis Sci 2019; 64(7): 1938–1944. doi: 10.1007/s10620-019-5474-4.
50. Liu W, Wang L, Muefong C et al. SPH3127 (sitokiren), a novel renin inhibitor, suppresses colitis development in mouse models of experimental colitis. Inflamm Bowel Dis 2025; 31(8): 2244–2253. doi: 10.1093/ibd/izaf097.
51. Sitaru S, Budke A, Bertini R et al. Therapeutic inhibition of CXCR1/2: where do we stand? Intern Emerg Med 2023; 18(6): 1647–1664. doi: 10.1007/s11739-023-03309-5.
52. Xv Y, Feng Y, Lin J. CXCR1 and CXCR2 are potential neutrophil extracellular trap-related treatment targets in ulcerative colitis: insights from Mendelian randomization, colocalization and transcriptomic analysis. Front Immunol 2024; 15: 1425363. doi: 10.3389/fimmu.2024.1425363.
53. Boyles JS, Beidler CB, Strifler BA et al. Discovery and characterization of a neutralizing pan-ELR+CXC chemokine monoclonal antibody. MAbs 2020; 12(1): 1831880. doi: 10.1080/19420862.2020.1831880.
54. Tinoco R, Otero DC, Takahashi AA et al. PSGL-1: a new player in the immune checkpoint landscape. Trends Immunol 2017; 38(5): 323–335. doi: 10.1016/j.it.2017.02.002.
55. Hope JL, Otero DC, Bae EA et al. PSGL-1 attenuates early TCR signaling to suppress CD8+ T cell progenitor differentiation and elicit terminal CD8+ T cell exhaustion. Cell Rep 2023; 42(5): 112436. doi: 10.1016/j.celrep.2023.112436.
56. Huang CC, Lu YF, Wen SN et al. A novel apoptosis-inducing anti-PSGL-1 antibody for T cell-meted diseases. Eur J Immunol 2005; 35(7): 2239–2249. doi: 10.1002/eji.200525849.
57. Lovewell RR, Langermann S, Flies DB. Immune inhibitory receptor agonist therapeutics. Front Immunol 2025; 16: 1566869. doi: 10.3389/fimmu.2025.1566869.
58. Chen Y, Ye X, Escames G et al. The NLRP3 inflammasome: contributions to inflammation-related diseases. Cell Mol Biol Lett 2023; 28(1): 51. doi: 10.1186/s11658-023-00462-9.
59. Weber S, Sitte S, Voegele A et al. NLRP3 inhibition leads to impaired mucosal fibroblast function in patients with inflammatory bowel diseases, J Crohns Colitis 2024; 18(3): 446–461. doi: 10.1093/ecco-jcc/jjad164.
60. Liu L, Dong Y, Ye M et al. The pathogenic role of NLRP3 inflammasome activation in inflammatory bowel diseases of both mice and humans. J Crohns Colitis 2017; 11(6): 737–750. doi: 10.1093/ecco-jcc/jjw219.
61. Parmar DV, Kansagra KA, Momin T et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of the oral NLRP3 inflammasome inhibitor ZYIL1: first-in-human phase 1 studies (single ascending dose and multiple ascending dose). Clin Pharmacol Drug Dev 2023; 12(2): 202–211. doi: 10.1002/cpdd.1162.
62. Watson N, Kuppuswamy S, Ledford WL et al. The role of HDAC3 in inflammation: mechanisms and therapeutic implications. Front Immunol 2024; 15: 1419685. doi: 10.3389/fimmu.2024.1419685.
63. Li C, Gu S, Zhang Y et al. Histone deacetylase in inflammatory bowel disease: novel insights. Therap Adv Gastroenterol 2025; 18: 17562848251318833. doi: 10.1177/17562848251318833.
64. Graßhoff H, Comdühr S, Monne LR et al. Low-dose IL-2 therapy in autoimmune and rheumatic diseases. Front Immunol 2021; 12: 648408. doi: 10.3389/fimmu.2021.648408.
65. Ko B, Takebe N, Andrews O et al. Rethinking oncologic treatment strategies with interleukin-2. Cells 2023; 12(9): 1316. doi: 10.3390/cells12091316.
66. Rosenzwajg M, Lorenzon R, Cacoub P et al. Immunological and clinical effects of low-dose interleukin-2 across 11 autoimmune diseases in a single, open clinical trial. Ann Rheum Dis 2019; 78(2): 209–217. doi: 10.1136/annrheumdis-2018-214229.
67. Allegretti JR, Mitsialis V, Canavan JB et al. Low-dose interleukin 2 for the treatment of moderate to severe ulcerative colitis. Gastroenterology 2023; 165(2): 492–495. doi: 10.1053/j.gastro.2023.03.230.
68. Deng Z, Fan T, Xiao C et al. TGF-β signaling in health, disease, and therapeutics. Signal Transduct Target Ther 2024; 9(1): 61. doi: 10.1038/s41392-024-01764-w.
69. Peng D, Fu M, Wang M et al. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol Cancer 2022; 21(1): 104. doi: 10.1186/s12943-022-01569-x.
70. Bertin L, Crepaldi M, Zanconato M et al. Advancing therapeutic frontiers: a pipeline of novel drugs for luminal and perianal Crohn‘s disease management. Therap Adv Gastroenterol 2024; 17: 17562848241303651. doi: 10.1177/17562848241303651.
71. Wu X, Sun M, Yang Z et al. The roles of CCR9/CCL25 in inflammation and inflammation-associated diseases. Front Cell Dev Biol 2021; 9: 686548. doi: 10.3389/fcell.2021.686548.
72. Wendt E, Keshav S. CCR9 antagonism: potential in the treatment of inflammatory bowel disease. Clin Exp Gastroenterol 2015; 8: 119–130. doi: 10.2147/CEG.S48305.
73. Kalindjian SB, Kadnur SV, Hewson CA et al. A new series of orally bioavailable chemokine receptor 9 (CCR9) antagonists; possible agents for the treatment of inflammatory bowel disease. J Med Chem 2016; 59(7): 3098–3111. doi: 10.1021/acs.jmedchem.5b01840.
74. Hedin CR, Creignou M, MacKay J et al. P1116 imaging of CCR9 in the small bowel of patients with Crohn’s disease: effects of the CCR9-depleting monoclonal antibody AZD7798. J Crohns Colitis 2025; (Suppl 1): i2048–i2049.