
Keywords
entecavir (ETV)
tenofovir disoproxil fumarate (TDF)
tenofovir alafenamide (TAF)
Abstract
The new recommendations reflect the increase in knowledge that has been reported since the release of previous Czech guidelines in September 2017. The base for these guidelines were European Association for the Study of the Liver (EASL) guidelines from May 2025. According to qualified estimates, there are 254 million people with chronic hepatitis B (HBV) infection worldwide. The Czech Republic is among the countries with a low prevalence of HBV infection. HBV infection can cause serious, life-threatening liver damage – fulminant hepatitis, liver cirrhosis, and hepatocellular carcinoma (HCC). The main goal of treatment is to prolong life and improve its quality by preventing the progression of chronic hepatitis to liver cirrhosis, decompensation of cirrhosis, and the development of HCC. A prerequisite for achieving this goal is sustained suppression of HBV replication. Other treatment goals include preventing vertical transmission of infection from mother to newborn, preventing HBV reactivation, treating extrahepatic manifestations (EHM) of HBV, and reducing the likelihood of HBV transmission between individuals. In general, there are two possible strategies for treating chronic hepatitis B: treatment with nucleos (t) ide analogues (NAs) or pegylated interferon alfa. Currently, the vast majority of patients in the Czech Republic and throughout Europe are treated with NAs. Entecavir (ETV), tenofovir disoproxil fumarate (TDF), or tenofovir alafenamide (TAF) should be used as first-line NAs. The main advantage of treatment with highly effective NAs with a high genetic barrier to resistance is predictably high and long-term antiviral efficacy leading to undetectable HBV DNA in serum in the vast majority of adherent patients and the favorable safety profile of these drugs. These NAs can be used to treat any patient with chronic hepatitis B and are the only treatment option for patients with decompensated liver cirrhosis, after liver transplantation, with EHM HBV infection, severe acute hepatitis B, or exacerbation of chronic hepatitis B. When deciding between ETV, TDF, and TAF, it is necessary to assess comorbidities (especially renal insufficiency and bone density reduction) and other factors (women of childbearing age, pregnant women, older age).
This article is an English translation of the original Czech publication. The translation was generated with the assistance of artificial intelligence and has been reviewed and approved by the authors. The original version remains the authoritative source.
Preamble
The recommendations for the diagnosis and treatment of hepatitis B virus (HBV) infection presented below were developed by members of the working groups on viral hepatitis of the Czech Society of Hepatology (ČHS), the Czech Medical Association of Jan Evangelista Purkyně (ČLS JEP), and the Society for Infectious Diseases (SIL) of the ČLS JEP. The new recommendations reflect the increase in knowledge published since the release of the previous ČHS and SIL guidelines in September 2017 [1,2]. The basis for drafting these guidelines was the European Association for the Study of the Liver (EASL) guidelines from May 2025 [3]. Maximum adherence to professional recommendations is a prerequisite for achieving effective care for patients with viral hepatitis B on a national scale. This document serves as a basis for negotiations between both professional societies and state authorities and healthcare payers.
HBV Epidemiology
According to qualified estimates by the World Health Organization (WHO), 254 million people worldwide were chronically infected with HBV in 2022. The annual incidence is estimated at 1.2 million new HBV infections. Approximately 1.08 million people die annually from the consequences of HBV infection, primarily from liver cirrhosis and hepatocellular carcinoma (HCC). It is projected that mortality associated with HBV infection will continue to rise from 858,000 in 2015 to 1.149 million in 2030. Specifically, deaths from HCC are projected to rise from 664,000 to 857,000 during those years, and deaths from decompensated cirrhosis from 296,000 to 403,000 [4]. The main problem is the low level of testing and poor access to treatment for chronic hepatitis B in many of the world’s poorer countries. According to a model developed in 2022 by the Polaris Observatory Collaborators, an international epidemiological initiative, the infection was diagnosed in only 36 million people with HBV infection, and only 6.8 million of the 83.3 million people indicated for treatment have been treated to date [5].
There are enormous regional differences in the prevalence of HBV infection – low prevalence is defined as hepatitis B surface antigen (HBsAg) positivity in less than 2% of the population, moderate prevalence is 2–8%, and high prevalence is more than 8% of those chronically infected with HBV. The Czech Republic is among the countries with low prevalence of HBV infection. According to the latest nationwide serological surveys from 2001, 0.56% of our citizens were chronically infected with HBV [6] . More recent results from the entire Czech Republic are not available. In 2013, a similar study was conducted in only two regions of the Czech Republic, and the prevalence found was only 0.064% [7]. In recent years, fewer than 50 cases of acute hepatitis B have been reported annually in our country; fluctuations are minimal, and a steady downward trend in cases of this serious disease can be observed. This is a result of long-term vaccination of a significant portion of our population against HBV. Due to population migration, there has been an increase in the number of reported cases of chronic hepatitis B in Czech Republic—127 cases in 2021, 244 in 2022, 380 in 2023, and 429 in 2024. Preliminary data for 2025 indicate a continuation of this upward trend—218 cases by the end of June 2025 [8].
The hepatitis B virus is detectable in most bodily fluids; therefore, transcutaneous and permucosal transmission play a major role in the spread of infection. In this regard, HBV is approximately 100 times more infectious than the human immunodeficiency virus (HIV) and 10 times more infectious than the hepatitis C virus (HCV). Globally, HBV infection is most commonly transmitted through sexual contact. In endemic regions (South and Southeast Asia, sub-Saharan Africa), vertical transmission of the infection from mother to newborn is of great significance. This transmission most commonly occurs during childbirth and breastfeeding. If the mother is highly viremic, HBV can also be transmitted transplacentally during the last trimester of pregnancy. There are still many countries where blood donors are not routinely tested for HBV infection, and in these less developed countries, transfusions of blood and blood products pose a real risk of HBV infection. A significant risk factor for HBV transmission is the sharing of injection equipment among intravenous drug users, although the incidence of HBV infection in this high-risk group is not as common as that of HCV infection [9].
Infections in newborns and young children are currently extremely rare in the Czech Republic thanks to screening of all pregnant women for HBsAg and subsequent passive and active immunization of newborns born to HBsAg-positive mothers. Furthermore, since 2001, universal vaccination of children against HBV has been carried out in our country—now starting at 9 weeks of age.
Natural course and clinical presentation of HBV infection
HBV infection is associated with a highly heterogeneous spectrum of liver diseases. Acute hepatitis B is predominantly a benign disease that resolves spontaneously in most cases; in 0.1–1% of cases, the disease takes a fulminant course with high mortality.
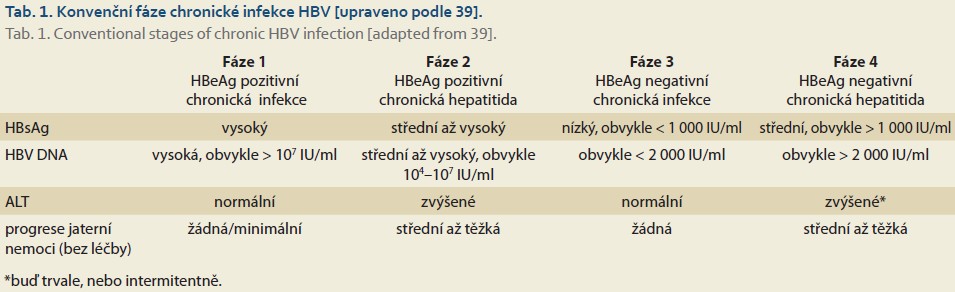
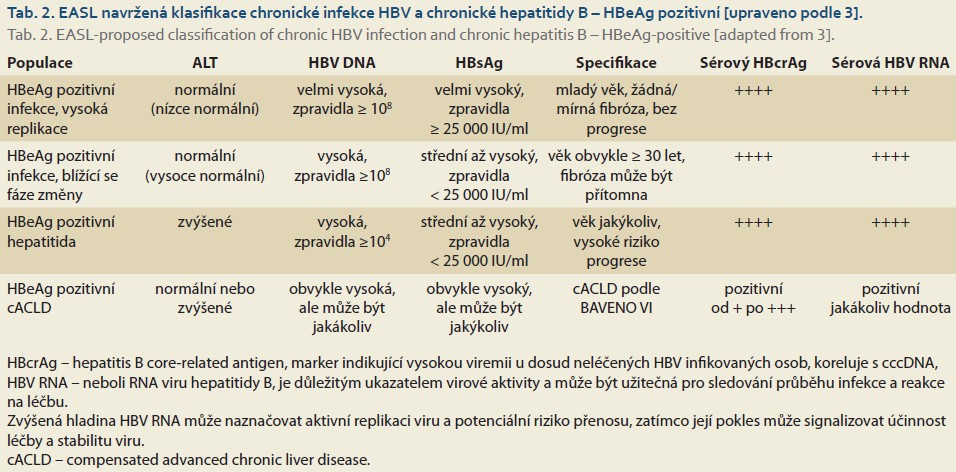
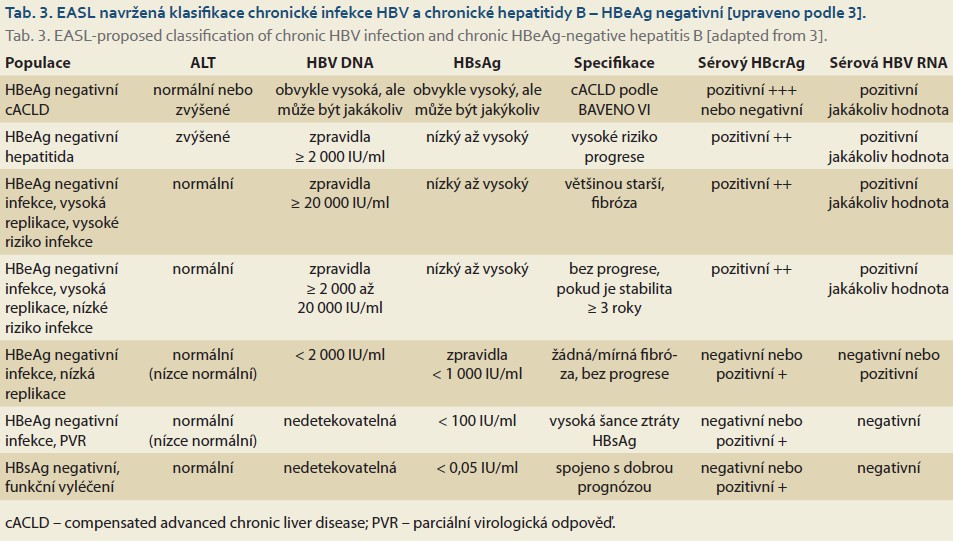
Chronic HBV infection refers to an infection lasting longer than 6 months. It is a dynamic process reflecting the interactions between viral replication and the host immune response. Not all patients chronically infected with HBV have chronic hepatitis B. The natural course of chronic HBV infection is schematically divided into four phases, based on the presence of the secretory antigen (HBeAg—hepatitis B “e” antigen), serum HBV DNA (HBV nucleic acid) concentrations, serum ALT (alanine aminotransferase) activity, and the likely progression of liver inflammation without antiviral treatment (Table 1). Current nomenclature is based on the description of two fundamental characteristics of chronicity—infection vs. hepatitis. Chronic HBV infection is a highly heterogeneous disease that often cannot be fully classified into the four traditional stages. This has led to the emergence of terms such as “grey zone” or “intermediate phase,” which have been used in studies to describe a patient population that cannot be fully classified into the classic categories [10–12]. The EASL recognizes that a simple and practical nomenclature for chronic HBV infection is essential for clinical practice, but in clinical research, a more precise classification of the individual phases of HBV infection is needed to capture the complex dynamics of this process. Therefore, 11 distinct patient categories have been proposed based on various viral markers, inflammatory activity, disease stage, and risk of progression (Tables 2, 3) [3].
Screening and Initial Evaluation of Patients with Chronic HBV Infection
Initial screening for HBV infection involves the detection of HBsAg and anti-HBc antibodies. When HBV infects hepatocytes, large amounts of HBsAg are released into the bloodstream, allowing for the detection of HBV infection even in its early stages. Highly sensitive enzyme-linked immunosorbent assays (ELISAs) with a lower limit of detection < 0.05 IU/ml (sometimes even < 0.005 IU/ml) are typically used to detect HBsAg. Nevertheless, false-negative HBsAg results may occur during the early stages of HBV infection. This window, during which HBsAg is negative and HBV DNA is positive, can last up to several weeks. Low concentrations of HBsAg may also be present in immunosuppressed patients [13].
The initial evaluation of a patient with newly diagnosed chronic HBV infection must include a complete medical history, physical examination, assessment of the activity and stage of liver disease, and serological markers of HBV infection. Sexual partners and household members of infected individuals should be advised to undergo testing for serological markers of HBV infection (HBsAg, total anti-HBc and IgM anti-HBc, anti-HBs). If these serological markers are negative, they should be vaccinated against HBV.
Quantitative testing for HBV DNA in serum is essential for diagnosing chronic HBV infection, determining its stage, deciding on treatment, and monitoring treatment efficacy. Highly sensitive polymerase chain reaction (PCR) is used to detect HBV DNA in serum. Results obtained by this method are reported in international units per milliliter (IU/ml). The lower detection limit of this method is 10–15 IU/ml [14].
Determination of HBeAg and anti-HBe is essential for determining the stage of chronic HBV infection.
All newly diagnosed HBsAg-positive individuals should undergo abdominal ultrasound, assessment of the stage of liver disease using non-invasive methods of liver stiffness measurement (LSM),
specifically transient elastography (VCTE – vibration -controlled transient elastography), or ultrasound-based methods (SWE – shear wave elastography) and (ARFI – acoustic ratio force impulse imaging), as well as blood tests (APRI – AST-platelet ratio index, FIB-4 – Fibrosis-4, FibroTest) [15].
The Baveno VI conference introduced the term “compensated advanced chronic liver disease – cACLD” for patients with advanced liver fibrosis or early cirrhosis who are asymptomatic but at risk of developing clinically significant portal hypertension (CSPH) and progression of liver disease. Liver stiffness < 10 kPa is a safe cutoff for ruling out cACLD, and stiffness > 15 kPa is highly suggestive of cACLD. In individuals with liver stiffness < 20 kPa and a platelet count > 150,000, the likelihood of developing high-risk varices and bleeding from them is low; therefore, preventive gastrofibroscopic examination is not necessary [16,17]. The VCTE-LSM cutoff values are listed in Table 4.

Liver biopsy is indicated in cases of diagnostic uncertainty—inconsistent results from non-invasive testing and the presence of liver comorbidities.
Quantitative determination of serum HBsAg concentration cannot replace HBV DNA quantification, but it provides additional important information regarding the determination of the stage of chronic HBV infection and, in particular, the assessment of the risk of discontinuing treatment before HBsAg clearance [18]. The significance of HBsAg quantification is evident from Table 5.

The determination of two new biomarkers, HBcrAg and HBV RNA, is gaining importance, as they allow for the assessment of the intrahepatic pool of covalently closed circular DNA (cccDNA – covalently closed circular DNA). The persistent presence of cccDNA in hepatocyte nuclei is the basis for reactivation and recurrence of HBV infection in immunocompromised individuals or during immunosuppressive therapy. Direct detection of cccDNA is possible only in a biopsy-obtained sample of liver tissue; therefore, the assessment of the following serological parameters is of great practical importance. HBcrAg—hepatitis B core-related antigen—is a marker indicating high viremia in previously untreated HBV-infected individuals; and its quantified value in the blood correlates well with the level of cccDNA in hepatocytes. HBV RNA—or hepatitis B virus ribonucleic acid— is an important indicator of viral activity and may be useful for monitoring the course of infection and response to treatment. Elevated HBV RNA levels may indicate active viral replication and a potential risk of transmission, while a decrease may signal treatment efficacy and viral stability [19–21].
Comorbidities must be systematically confirmed or ruled out, including alcoholic, autoimmune, and metabolic liver disease, as well as coinfection with hepatitis D virus (HDV), hepatitis C virus (HCV), and HIV.
Testing for the presence of antibodies against hepatitis A virus (HAV) should be performed, and patients with negative anti-HAV IgG antibodies should be vaccinated against HAV.
Treatment Goals for Chronic Hepatitis B Virus Infection
The primary goal of treatment is to prolong life expectancy and improve quality of life by preventing the progression of chronic hepatitis to liver cirrhosis, decompensated cirrhosis, liver failure, and the development of HCC, and by extending life expectancy [3].
Because the achievement of the main treatment goals can only be assessed after many years, other goals have been defined that allow the success of treatment to be assessed earlier:
- Achieving long-term suppression of serum HBV DNA levels, ideally below the lower limit of PCR sensitivity (10–15 IU/ml), is the primary objective of current therapeutic strategies.
- The ideal treatment outcome is sustained HBsAg clearance with or without seroconversion to anti-HBs. This is an indicator of deep suppression of HBV replication and viral protein expression.
- ALT normalization is a secondary goal.
Additional goals of antiviral therapy include:
- In HBeAg-positive patients, a satisfactory treatment outcome is considered to be loss of HBeAg with or seroconversion to anti-HBe, combined with a decrease in HBV DNA < 2,000 IU/ml, which often represents partial immune control of chronic HBV infection.
- Reduction in the stage of liver fibrosis.
- Improvement in extrahepatic manifestations (EHM) of HBV infection.
- Improved quality of life.
- Prevention of HBV transmission.
- Prevention of HBV reactivation and recurrence.
Evolving treatment goals for chronic HBV infection with nucleos(t)ide analogues
Even many years of treatment with nucleos(t)ide analogues (NA) does not lead to the achievement of all current treatment goals in the vast majority of individuals with chronic HBV infection. Therefore, efforts are being made to replace the maximalist goal of complete cure—with the eradication of all signs of HBV infection from the liver, including cccDNA and integrated HBV DNA— a more realistic goal known as functional cure (sustained loss of HBsAg, ideally with anti-HBs positivity, undetectable HBV DNA). This is related to the growing effort to discontinue treatment of chronic HBV infection before achieving a complete cure, which is unrealistic with currently available treatments.
The significance of HBV DNA suppression
- An HBV DNA concentration ≥ 2,000 IU/ml is associated with an increased risk of liver cirrhosis and HCC [22,23].
- The relationship between serum HBV DNA concentration and the risk of HCC is not linear.
- The risk of HCC is significantly higher at HBV DNA > 200,000 IU/ml compared to the risk at HBV DNA concentrations between 2,000 IU/ml and 200,000 IU/ml.
- Conversely, the risk of HCC is very low when HBV DNA is < 2,000 IU/ml [24,25].
General indications for the treatment of chronic hepatitis B
- In principle, all HBsAg-positive patients with detectable HBV DNA are candidates for antiviral therapy. The indication for treatment is primarily based on an assessment of HBV DNA concentration, ALT activity, fibrosis stage, and the risk of liver disease progression and HCC development.
- All patients with both HBeAg-positive and HBeAg-negative chronic hepatitis B, defined by serum HBV DNA concentration > 2,000 IU/ml, ALT > upper limit of normal (ULN), and/or at least moderate inflammatory-necrotic process or fibrosis, should be treated.
- Patients with liver cirrhosis or advanced liver fibrosis (METAVIR score ≥ 3 on liver histology or LSM > 8 kPa) should be treated if HBV DNA is detectable in their serum (at any concentration), regardless of ALT activity.
- Patients with persistently low HBV DNA (< 2,000 IU/mL) and persistently elevated ALT levels (> ULN) should be treated. However, it is necessary to determine whether the ALT elevation is not caused by another liver disease.
- Individuals with HBeAg-positive or HBeAg-negative chronic HBV infection require individual assessment to determine indications for antiviral therapy [3].
Monitoring of Untreated Patients
Patients who are not candidates for immediate antiviral therapy should be monitored for ALT activity, serum HBV DNA levels, and fibrosis stage (using non-invasive methods) :
- Individuals with newly diagnosed HBsAg positivity should be monitored during the first year following diagnosis, or until the initiation of antiviral therapy, with HBV DNA and ALT testing every 3–6 months. After this initial phase, the intervals between follow-up visits may be extended to 6–12 months depending on the stage of the disease.
- Quantitative HBsAg testing should be performed every 12 months. If quantitative HBsAg testing is not available, qualitative HBsAg detection is the minimum requirement.
- HBeAg-positive patients should be tested for the presence of HBeAg and anti-HBe every 12 months and at any significant change in ALT activity.
- The frequency of non-invasive methods assessing the progression of liver fibrosis depends on the stage of the disease and the presence of comorbidities (individual assessment of need) [3].
Treatment of patients with HBeAg-positive chronic HBV infection
A) In young patients (< 30 years of age) who are HBeAg-positive, with persistently normal ALT levels, no significant fibrosis, no family history of HCC, and no immunosuppressive therapy:
- Current clinical evidence does not support the necessity of immediate antiviral treatment.
- On the other hand, such early treatment has potential benefits in reducing HBV DNA integration and clonal expansion, given the high viremia in these individuals.
B) Treatment is indicated for HBeAg-positive chronic infection:
- In cases of increased risk of HCC development.
- In immunocompromised individuals or those on immunosuppressive therapy to prevent the development of hepatitis.
- In selected individuals with an increased risk of transmitting the infection to others.
- In pregnant women with HBV DNA ≥ 200,000 IU/ml to prevent vertical transmission [3].
Treatment of patients with HBeAg-negative chronic HBV infection
The absence of HBeAg is generally associated with lower HBV DNA concentrations and a consequent approximately 10-fold lower risk of HBV transmission, although the risk of person-to-person transmission cannot be completely ruled out. Therefore, in some countries, treatment is indicated for healthcare workers for epidemiological reasons to maintain their HBV DNA concentration below 2,000 IU/ml or even below 200 IU/ml if they perform invasive procedures [26,27]. In the Czech Republic, this requirement is not legally mandated, but it cannot be assumed that this would pose a significant problem—long-term vaccination of healthcare workers prior to entering medical schools or nursing colleges, widespread vaccination of children since 2001, which also included current young doctors and nurses, and the very low proportion of foreigners from HBV-endemic regions among our healthcare workers.
A) Patients with HBeAg-negative chronic infection (HBV DNA consistently < 2,000 IU/ml, persistently normal ALT levels, no signs of liver fibrosis) have a low risk of disease progression and transmission of HBV infection to another person; therefore, they usually do not require immediate antiviral therapy.
B) Treatment is indicated for HBeAg-negative chronic infection:
- In cases of increased risk of HCC development.
- In immunocompromised individuals or those on immunosuppressive therapy to prevent the development of hepatitis.
- In selected individuals with an increased risk of transmitting the infection to another person.
- In pregnant women with HBV DNA ≥ 200,000 IU/ml to prevent vertical transmission [3].
Treatment strategies for chronic hepatitis B
Generally, two treatment strategies for chronic hepatitis B are available—treatment with NA or pegylated interferon-α (PEG-IFN). In the Czech Republic, PEG-IFN treatment is practically never used because there are not enough suitable patients for this therapy, which has a number of serious adverse effects and numerous contraindications. Therefore, we will not discuss it in detail in the following text. The 2025 EASL Guidelines devote sufficient attention to it [3].
The main advantage of treatment with highly effective NAs with a high genetic barrier to resistance (ETV – entecavir, TDF – tenofovir disoproxil fumarate, TAF – tenofovir alafenamide) is their predictably high and long-term antiviral efficacy, leading to undetectable serum HBV DNA in the vast majority of adherent patients, and the favorable safety profile of these drugs. These NAs can be used to treat any patient with chronic hepatitis B and represent the only treatment option for patients with decompensated liver cirrhosis , after liver transplantation, with EHM, acute hepatitis B, or severe exacerbation of chronic hepatitis B. Only NAs can be used to prevent HBV reactivation during immunosuppression and to prevent HBV transmission from patients with high viremia who do not meet the typical indication criteria for treatment.
- ETV, TDF, or TAF should be used as first-line NA treatment.
- When deciding between ETV, TDF, and TAF, comorbidities (primarily renal insufficiency and reduced bone density) and other factors (women of childbearing age, pregnant women, older age) must be evaluated (Table 6) .
- HBV DNA suppression is comparable with ETV, TDF, or TAF treatment—according to clinical trial results, 90–100% after 1–10 years of treatment [3].
The general treatment algorithm for chronic HBV infection is shown in Scheme 1.

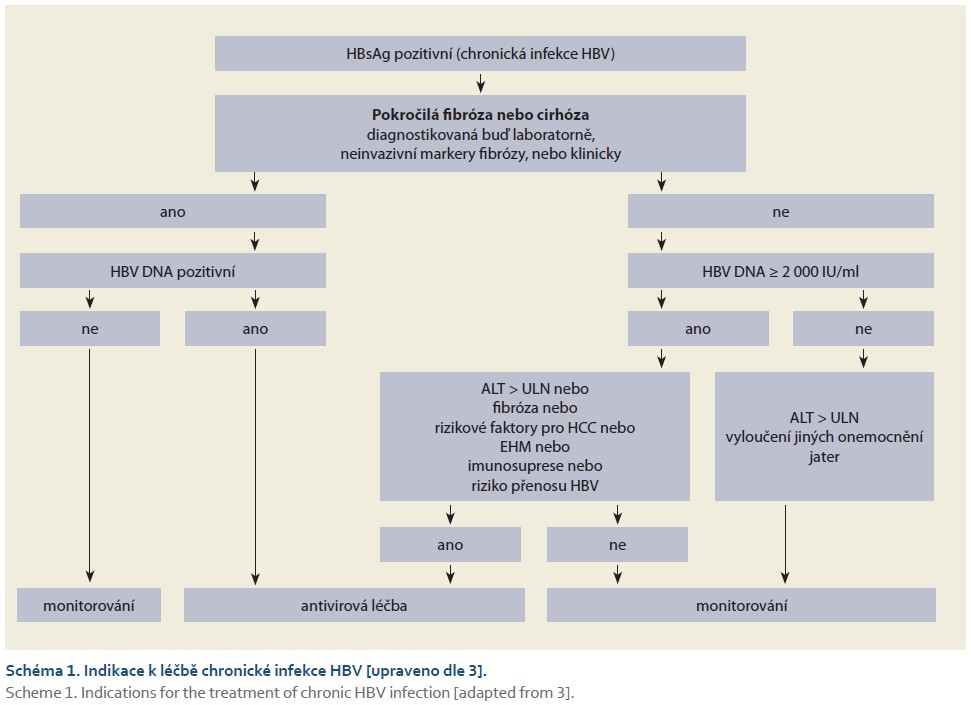
Definition of response to antiviral treatment
- Complete virological response (CVR) – undetectable HBV DNA using a sensitive assay (< 20 IU/ml).
- Partial virological response (PVR) – viremia does not decline steadily and remains > 2,000 IU/ml.
- Virological non-response (VNR) – HBV DNA decrease < 1 log10 after 6 months of treatment.
- Virological resistance (VR) – HBV DNA increase ≥ 1 log10 from the nadir [3].
Monitoring of patients treated with ETV, TDF, or TAF
- During treatment, HBV DNA and ALT should be monitored every 3–6 months until virologic response is achieved; thereafter, the monitoring interval may be extended to 6–12 months when treating with ETV, TDF, or TAF.
- HBsAg testing every 12 months, preferably quantitatively.
- For HBeAg-positive patients, HBeAg/anti-HBe testing every 12 months.
- Assess renal function before treatment and regularly during treatment.
- Switch from TDF to TAF or ETV in the event of a decrease in glomerular filtration rate (GFR), the development of tubulopathy, hypophosphatemia, or osteoporosis.
- Non-invasive fibrosis assessment every 12–24 months [3].
In both HIV- and HBV-positive patients, many studies have demonstrated higher lipid levels with TAF treatment compared to TDF. Real-world data showed that switching from TDF to a TAF regimen (without other medication changes) led to a gradual rise in LDL-cholesterol levels lasting longer than 9 months, while triglyceride (TGL) levels rose for 9–16 months before stabilizing. Weight gain following the switch from TDF to TAF is also well documented and may contribute to changes in the lipid profile. In patients with chronic HBV infection, pre-existing diabetes mellitus and hypertension were identified as risk factors for worsening lipid profiles during TAF treatment. Conversely, several studies have shown that TDF has a mild effect on lowering lipid levels. These facts should be taken into account when considering switching from TDF to TAF, particularly in patients with pre-existing metabolic risk and to monitor lipid levels in patients treated with TAF [28–32].
Management in the event of a partial virologic response (PVR), virologic non-response (VNR), or the development of resistance (VR) during NA
- In the case of PVR or VNR, the first step is to check adherence.
- If adherence is adequate, HBV resistance testing is warranted.
- Management in cases of confirmed PVR, VNR, or VR:
a) switch from a nucleoside analog (LAM – lamivudine, ETV) to TAF or TDF;
b) switch from TDF or TAF to ETV or a combination of ETV with TDF or TAF. - In the event of persistent low viremia (< 2,000 IU/ml) or the appearance of a short-term increase in HBV DNA (“blip”) during treatment with TDF, TAF, or ETV, it is not necessary to immediately change treatment unless advanced fibrosis is present and resistance has been ruled out—poor adherence or reduced intestinal absorption may explain the blip.
- In patients with liver cirrhosis, the goal is to achieve undetectable HBV DNA, ideally within 12 months of NA treatment. If this is not achieved, a change in treatment is warranted [3].
Resistance arising during treatment with ETV, TDF, or TAF is very rare. These NAs have a high genetic barrier against the development of resistance even during long-term treatment. This is why their use in first-line therapy is so important, as the development of resistance typically manifests clinically as hepatitis B reactivation, which is associated with higher morbidity and mortality, particularly in patients with cirrhosis and advanced liver fibrosis [33].
Genotypic resistance to ETV is rare in previously untreated patients—1.2% over 5 years of treatment. In patients previously treated with LAM, the likelihood of rapid resistance development is much higher – 6% after 1 year, more than 50% after 5 years. Therefore, ETV should not be used at all in the treatment of patients with proven resistance to LAM, and following prior treatment with LAM, a double dose of ETV is used, i.e., 1 mg daily [34,35].
The likelihood of developing resistance to TDF or TAF is extremely low – after 5 years of treatment, it has not been demonstrated for either tenofovir-containing formulation [36,37].
When can NA treatment be discontinued?
General options for discontinuing NA treatment
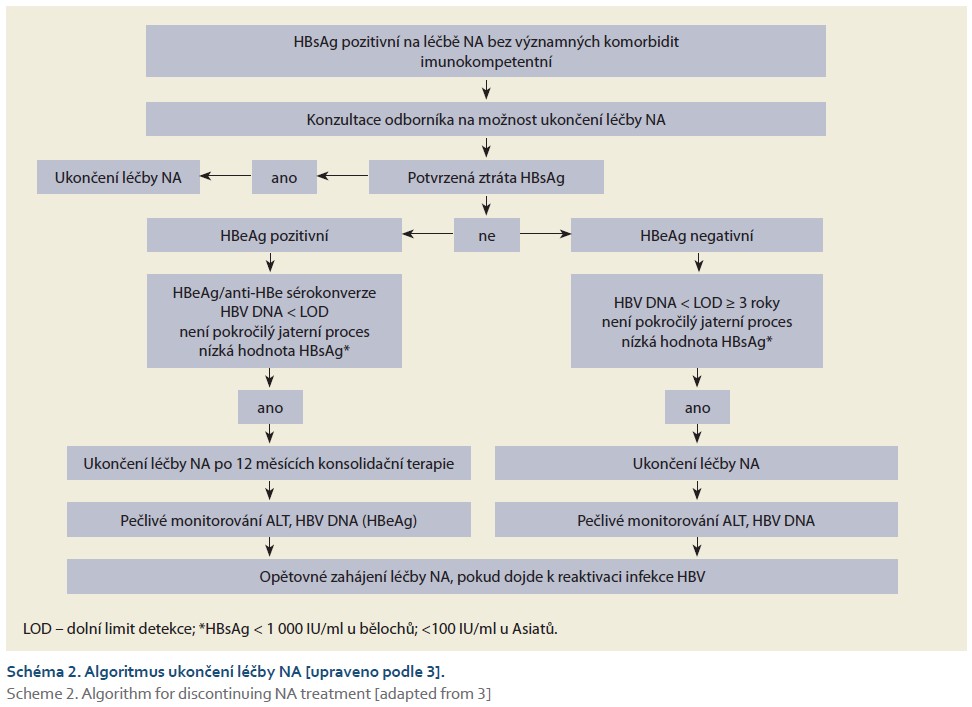
The general algorithm for discontinuing treatment of chronic HBV infection is shown in Figure 2.
- Discontinuing NA treatment is a very difficult and responsible decision that requires extensive experience on the part of the physician who prescribes and discontinues antiviral therapy.
- It is necessary to assess in advance the HBeAg status, HBsAg level, duration of HBV DNA suppression, degree of liver fibrosis, comorbidities, and the patient’s preferences and understanding of the possible consequences of discontinuing treatment.
- After confirmed loss of HBsAg (with or without anti-HBs seroconversion) and in the absence of risk factors, NA treatment may be discontinued.
- Quantification of HBsAg is important when deciding to discontinue NA treatment—a low HBsAg level is defined as < 1,000 IU/ml in Caucasians and < 100 IU/ml in Asians (more robust data are available from HBeAg-negative patients) .
- In initially HBeAg-positive individuals without signs of advanced liver disease, NA treatment can be discontinued 12 months after confirmed HBeAg/anti-HBe seroconversion and HBV DNA negativity – careful monitoring is necessary after treatment discontinuation.
- In selected HBeAg-negative patients without advanced liver disease, treatment may be discontinued prior to HBsAg loss if HBV DNA has been undetectable for 3–4 years and HBsAg levels are low—careful monitoring is necessary after treatment discontinuation.
- In addition to assessing HBsAg levels quantitatively, knowledge of HBcrAg and HBV RNA concentrations may aid in the decision to discontinue treatment—if these tests are available [3].
Discontinuation of NA treatment after HBsAg loss
- HBsAg loss is very rare with currently available NA treatment (usually around 1% per year).
- If HBsAg seroconversion occurs, it may indicate effective control of HBV infection, which is associated with the best long-term prognosis.
- After HBsAg loss, there is a possibility of seroreversion (recurrence of HBsAg positivity) due to the continued persistence of cccDNA in the liver.
- If HBsAg loss is repeatedly confirmed in tests 6 months apart, the risk of seroreversion is very low.
- After HBsAg loss, anti-HBs antibodies appear in only a small number of treated NA patients; therefore, the criteria for discontinuing NA treatment are primarily based on HBsAg loss.
- Post-treatment monitoring: HBV DNA and ALT every 3 months for the first year, then every 6–12 months, also in view of HCC surveillance.
- In patients with compensated liver cirrhosis, treatment may be discontinued only after confirmed HBsAg/anti-HBs seroconversion or after 12 months of consolidation therapy following HBsAg loss.
- The main concern is that potential HBsAg seroreversion associated with an increase in HBV DNA may lead to liver decompensation.
- In patients with decompensated cirrhosis, treatment may be discontinued only after confirmed HBsAg/anti-HBs seroconversion; according to some authors, treatment should never be discontinued [3].
Discontinuation of NA treatment prior to HBsAg loss in HBeAg-positive patients
- In initially HBeAg-positive individuals without signs of advanced liver disease, NA treatment may be discontinued 12 months after confirmed HBeAg/anti-HBe seroconversion and HBV DNA negativity—careful monitoring is necessary after treatment discontinuation.
- However, most data come from Asian patients, so it is not entirely clear whether this also applies to the Caucasian population.
- Heterogeneous data suggest that a lower HBsAg level at the time of NA discontinuation increases the likelihood of a sustained response (in Caucasians < 1,000 IU/ml, in Asians < 100 IU/ml—more robust data are available for HBeAg-negative individuals).
- Discontinuation of NA treatment in individuals with persistent HBeAg positivity is not recommended even with long-term HBV DNA negativity [3].
Discontinuation of NA treatment prior to HBsAg loss in HBeAg-negative patients
- In selected HBeAg-negative patients without advanced liver disease, treatment may be discontinued prior to HBsAg loss if HBV DNA has been undetectable for 3–4 years and HBsAg levels are low—careful monitoring is necessary after treatment discontinuation.
- In cases of advanced liver disease, NA treatment should not be discontinued before HBsAg loss, ideally after HBsAg/anti-HBs seroconversion [3].
Risk of HCC development in individuals with chronic HBV infection
Risk factors associated with HCC development in individuals with chronic HBV infection can be divided into viral, host, and environmental factors (Tables 7–10). Recommendations for inclusion in the HCC surveillance program are provided in Table 11.
- Liver cirrhosis is the most significant risk factor for HCC; even after viral suppression and possible loss of HBsAg, the risk of developing HCC persists.
- Abdominal ultrasound should be performed every 6 months in all at-risk patients—it is important that the procedure be performed by an experienced sonographer.
- Other imaging methods (contrast-enhanced CT – computed tomography, MRI – magnetic resonance imaging) are indicated if the ultrasound result is unreliable.
- Tumor markers (e.g., AFP – alpha-1-fetoprotein) can be used in conjunction with imaging tests; this may increase the sensitivity of detecting early-stage HCC, but 10–20% of HCC cases are not associated with elevated AFP [3].





Treatment of infection in patients with HCC
- HBsAg-positive patients with HCC should be treated with NA regardless of HBV DNA levels.
- TDF is considered the preferred drug for tertiary prophylaxis following curative HCC therapy (resection, locoablative therapy, liver transplantation) – significantly lower risk of tumor recurrence and improved survival compared to ETV (there is currently insufficient data for the use of TAF in this indication).
- NA treatment is also indicated in patients with initially unresectable HCC—slowing of tumor progression, improved survival [3].
Should acute HBV infection be treated with antivirals?
There is a very high probability of HBsAg loss (>95%) during the acute phase of HBV infection; therefore, it is unlikely that antiviral treatment would further increase the rate of HBsAg loss, and this has not yet been documented in the literature [39].
- Antiviral treatment for NA is indicated only in cases of impaired liver synthetic function, and collaboration with a transplant center is recommended to ensure proper timing of liver transplantation, if necessary
- The duration of NA treatment is until HBsAg loss.
- Most experience has been gained with LAM, but recent data show that ETV, TDF, or TAF are at least as effective and safe [3].
Available data have not demonstrated that antiviral treatment of severe cases of acute hepatitis B (INR < 1.5) provides any benefit in terms of improved survival or reduced need for liver transplantation [40]. Therefore, antiviral treatment of acute hepatitis B is generally not recommended at this time [41]. There is also a possible adverse effect of early NA treatment on the suppression of the HBV-specific immune response and a reduced likelihood of HBsAg loss, although there is currently no consensus on this issue [40,42].
Treatment of HBsAg-positive pregnant women
According to the US FDA Pharmaceutical Pregnancy Categories, which distinguishes five categories of drugs (A, B, C, D, X) based on potential effects on pregnancy, tenofovir and telbivudine (TBV) fall into Category B, meaning that although no adverse effects on the fetus have been reported in animal studies, controlled human studies are lacking, which is understandable. LAM, ADV, and ETV are classified as Category C — adverse effects on the fetus have been reported in animal studies. However, there is sufficient clinical data from clinical trials and large registries of pregnant women to support the view that LAM, TBV, ETV, and TAF are safe during pregnancy. Even when administered during the first trimester of pregnancy, no increased risk of fetal malformations has been reported. Most information on NA safety comes from HIV-positive women treated with drugs effective against both HIV and HBV infection. Given their efficacy and resistance-saving properties, TDF and TAF are the first-line drugs during pregnancy and for preventing vertical transmission of HBV infection [43–49].
A) Existing treatment with TDF or TAF should continue during pregnancy; treatment with ETV or ADV (and likely LAM) should be switched to TDF or TAF.
B) Indications for initiating antiviral treatment during pregnancy:
- chronic hepatitis B with general treatment recommendations;
- HBV DNA ≥ 200,000 IU/ml – prevention of vertical transmission.
C) Treatment with TDF or TAF to prevent vertical transmission should ideally be initiated before the start of the third trimester of pregnancy.
D) This treatment should continue after delivery to suppress viral replication.
E) Women treated with TDF or TAF may breastfeed.
Transplacental transmission of HBV can occur in highly viremic mothers despite passive and active immunization of the newborn. This risk rises to as high as 30% if HBV DNA in the pregnant woman exceeds 6–8 log10 IU/ml [50,51]. This risk can be reduced by rapidly lowering viremia to < 200,000
IU/ml with NA treatment (preferably tenofovir) [43,44]. Antiviral treatment can be initiated at any stage of pregnancy, including the first trimester. However, most data come from studies in which NA treatment began between the 28th and 32nd weeks of pregnancy [43,44,51].
To date, no case has been reported of HBV transmission from a mother with viremia < 200,000 IU/ml if the newborn was properly passively and actively immunized. Therefore, antiviral treatment is not indicated in pregnant women with HBV DNA < 200,000 IU/ml if the newborn’s immunization is ensured [43,51].
The decision to continue NA treatment after delivery depends on a number of factors. If the woman is planning another pregnancy, has chronic hepatitis or fibrosis, or wishes to continue antiviral treatment, NA treatment should continue. If treatment was initiated to prevent vertical transmission and there are no general indications for treating HBV infection, NA treatment may be discontinued shortly after delivery (according to Chinese recommendations, after 1–3 months). However, monitoring of HBV DNA and ALT is necessary thereafter. The exact duration of NA administration postpartum for preventive reasons has not yet been clearly established. Discontinuing NA immediately after delivery likely does not significantly increase the risk of virological relapse in the mother compared to continuing treatment for 4–8 weeks postpartum [52] .
The concentrations of TAF, TDF, and LAM in breast milk are very low, so the newborn’s exposure to TDF during breastfeeding is lower than in utero. According to another study, TAF was not detected in breast milk at all, and TDF was found in very low concentrations in both milk and umbilical cord blood. According to current knowledge, breastfeeding is possible even when the mother is being treated with TAF or TDF [53–56].
The issue of reducing the risk of vertical transmission of HBV infection via cesarean section has not yet been definitively resolved. A cesarean section is not routinely indicated provided that proper passive and active immunization of the newborn is ensured as soon as possible after birth. If the mother’s HBV DNA is ≥ 200,000 IU/ml at the time of delivery, a cesarean section may be considered following a discussion with the patient in which all arguments for and against such a decision are explained to her in detail [57–61].
Treatment of patients with decompensated liver cirrhosis and acute-on-chronic liver failure (ACLF)
- Patients with decompensated liver cirrhosis or ACLF and detectable HBV DNA in serum (regardless of its level) require urgent initiation of antiviral therapy with ETV, TDF, or TAF in specialized centers affiliated with transplant units [3].
NA therapy is highly effective and safe in patients with decompensated liver cirrhosis and leads to clinical improvement—a reduction in the risk of HCC, a decrease in the MELD score, and a lower Child-Pugh score. Treatment often leads to stabilization of the condition—more than 80% of patients survive without the need for liver transplantation, and up to one-third of initially decompensated cirrhotic patients achieve compensation [62,63]. In patients with ACLF (acute liver failure superimposed on chronic liver disease), NA treatment also improves survival, liver function, and virologic response [64–66].
In patients with both compensated and decompensated liver cirrhosis, the goal is to achieve undetectable HBV DNA, ideally within 12 months of NA treatment. When using ETV, TDF, or TAF,
achieving this goal is highly likely. In the past, the recommended dose of ETV for patients with decompensated cirrhosis was 1 mg daily, as it was assumed that some of these patients were infected with an HBV mutant resistant to LAM. More recent studies consider a daily dose of 0.5 mg to be sufficient even in patients with decompensated cirrhosis [63].
A commonly reported potential adverse effect of NA therapy in patients with decompensated cirrhosis is the development of lactic acidosis. For this reason, careful monitoring is necessary, especially in patients with a MELD score > 22 and impaired renal function.
Treatment of HBV/HDV Coinfection
Complete recommendations for the treatment of chronic hepatitis D are included in the EASL Clinical Practice Guidelines on Hepatitis D [67] from 2023 and in the DP ČHS and SIL from the same year [68,69].
- NA treatment should be administered to both compensated and decompensated cirrhotic patients.
- Patients without liver cirrhosis with HBV DNA ≥ 2,000 IU/ml should be treated with NA [3].
According to new findings published since the release of the aforementioned guidelines for HDV treatment, treatment with bulevirtide (BLV) does not worsen liver function in patients with liver cirrhosis and portal hypertension. In some cases of advanced, but not decompensated cirrhosis, an improvement in liver function has been observed with BLV treatment. The greatest risk is rebound HDV RNA after discontinuation of BLV treatment, particularly in decompensated patients, which may cause further deterioration of liver function [70,71] . The use of BLV in patients with decompensated liver disease is currently still off-label.
Treatment of HBV/HIV Coinfection
An estimated 3 million people worldwide are coinfected with HBV and HIV [72]. An increased risk of liver fibrosis progression and HCC development has been demonstrated in this coinfection—the risk of HCC increases in a time-dependent manner starting at HBV DNA ≥ 200 IU/ml [73].
- HBsAg-positive individuals with HIV infection should be treated with NAAs regardless of HBV DNA levels and ALT.
- Antiretroviral therapy (ART) should include TAF or TDF; if these drugs are contraindicated or unavailable, ETV may be administered concurrently with fully suppressive ART.
- Treatment monitoring and any necessary adjustments follow the same guidelines as for treatment of HBV monoinfected patients, taking into account the concurrent HIV infection.
- ART containing TAF or TDF should not be discontinued due to the risk of virological and biochemical relapse following the end of NA treatment [3].
Treatment of HBV/HCV Coinfection
Studies have shown that HBV/HCV coinfection worsens the prognosis of liver disease compared to HBV or HCV monoinfection, including a significantly higher incidence of HCC [74,75].
- Chronic infection in HBsAg-positive patients is treated with direct-acting antivirals (DAAs)—treatment efficacy for both HCV and HBV is high and comparable to that for monoinfections.
- All HBsAg-positive patients with liver cirrhosis should receive NA during DAA treatment to prevent HBV reactivation, even if they are HBV DNA-negative.
- Prophylactic NA administration is necessary during hepatitis C treatment with DAA, even if the patient does not meet the criteria for treatment of chronic HBV infection—e.g., HBV DNA < 2,000 IU/ml, normal ALT, absence of advanced fibrosis/cirrhosis [3].
HBV reactivation occurs during DAA treatment or shortly after its completion. Rarely, a severe flare of hepatitis activity occurs during HBV reactivation, sometimes leading to liver failure requiring a liver transplant. The risk of HBV reactivation ranges from 5.9% to 24%; flares are less common—2–9%. Higher baseline HBsAg and HBV DNA levels increase the risk of HBV reactivation, while NA prophylaxis significantly reduces this risk—this is particularly important in patients with cirrhosis. The duration of NA prophylaxis should be at least 12 weeks after the end of DAA treatment. In HBsAg-negative and anti-HBc-positive individuals, the risk of HBV reactivation during DAA treatment is low – 0.16–2%; reactivation is often transient and is usually not accompanied by a hepatitis “flare” or HBsAg seroconversion – therefore, routine NA prophylaxis is not recommended for these individuals, with the exception of those with cirrhosis [76–78].
What are the options for antiviral treatment in children and adolescents?
The following can be used for the treatment of children and adolescents:
- Interferon (IFN) α-2b is approved by the European Medicines Agency (EMA) and the U.S. FDA for children aged 1 year and older.
- PEG-IFN-α-2a for children aged 3 years and older.
- LAM for children aged 3 years and older.
- ETV for children aged 2 years and older.
- TDF is approved by the EMA for children aged 2 years and older, and by the FDA for those aged 12 years and older.
- TAF is approved by the EMA for children aged 12 years and older and for adults of any age weighing more than 35 kg [3].
Prophylaxis of HBV Reactivation and Recurrence
A summary of recommendations for the prophylaxis of HBV reactivation and recurrence during immunosuppressive (IS) therapy is provided in Tables 11–14.
- Testing for the presence of HBV markers (HBsAg, anti-HBc, HBV DNA) is always required prior to initiating IS therapy.
- HBsAg-positive individuals starting IS therapy should undergo the same tests as all other HBsAg-positive individuals.
- HBsAg-negative and anti-HBc-positive individuals should be tested for HBV DNA before starting IS therapy.
- HBsAg-positive patients at high and moderate risk of reactivation should receive prophylactic NA treatment.
- HBsAg-positive individuals at low risk of reactivation do not need to receive immediate NA prophylaxis, but their HBV DNA must be monitored at least every 3 months.
- HBsAg-negative, anti-HBc-positive, and HBV DNA-positive individuals—follow the same procedure as for HBsAg-positive individuals.
- HBsAg-negative, anti-HBc-positive, and HBV DNA-negative individuals should receive prophylactic NA if they are at high risk of HBV reactivation due to IS treatment.
- HBsAg-negative, anti-HBc-positive, and HBV DNA-negative individuals who are at moderate or low risk of reactivation due to IS treatment do not need to receive NA prophylaxis immediately, but their HBsAg and/or HBV DNA must be monitored every 3 months [3].



The following recommendations apply to all individuals requiring prophylactic NA administration.
- Generally, ETV, TDF, or TAF should be used for HBV reactivation prophylaxis.
- The duration of NA prophylaxis is not clearly defined, but it should last at least 6–12 months after the end of IS therapy.
- In the case of IS therapy leading to B-cell depletion, NA prophylaxis should last at least 18 months after the end of IS therapy.
- Ideally, discontinuation of prophylactic NA administration should meet the general criteria for discontinuing NA therapy. This applies particularly if HBV DNA is positive prior to initiating NA prophylaxis.
HBV replication alone does not lead to the destruction of infected hepatocytes; in most cases, the virus is not cytopathogenic. While immunosuppressive therapy reduces the immune response directed against infected hepatocytes, it also weakens immune control of viral replication.
The proportion of infected hepatocytes increases, HBV replication rises significantly, and reaches a level where it leads to direct damage to infected hepatocytes, thereby rendering HBV cytopathogenic. The clinical correlate of these processes is the development of fibrosing cholestatic hepatitis, a serious life-threatening disease that, without effective treatment, leads to liver failure in most cases within a few weeks. The condition in which immunosuppressive therapy leads to a significant increase in HBV replication and a flare-up of hepatitis in a previously inactive HBsAg carrier is referred to as reactivation of chronic hepatitis B. Recurrence refers to a condition in which a patient who has had hepatitis B and whose serum HBsAg has cleared becomes HBsAg-positive again and HBV replication rapidly increases. This is because viral nucleic acid persists permanently in hepatocytes in the form of so-called cccDNA (covalently closed circular DNA), even in patients who have become HBsAg-negative. Complete elimination of HBV never occurs. The condition previously referred to as viral elimination is merely effective immune control of replication, with viremia dropping below the detection limit. Loss of immune control over viral replication due to immunosuppressive therapy thus leads to recurrence of hepatitis B. The 2025 EASL Guidelines [3] use only the term “reactivation” and does not use the term “recurrence.” According to the EASL, reactivation refers to a state characterized either by renewed HBV DNA positivity (> 100 IU/ml) or seroreversion (HBsAg positivity following a period of negativity), or at least a 10-fold increase in HBV DNA levels from baseline. The Czech authors consider the distinction between reactivation and recurrence to be useful. We also believe that if replication cannot be adequately monitored during IS treatment, prophylaxis should be administered, particularly in HBsAg-positive individuals, even though the risk of reactivation is relatively low according to EASL recommendations. Reactivation and recurrence of HBV infection are potentially life-threatening situations; therefore, we consider their prophylaxis a priority, particularly during or after IS therapy. The risk of their occurrence is quantified into 3 levels: high (> 10%), moderate (1–10%), and low (< 1%). Reactivation occurs in 15–50% of inactive HBsAg-positive individuals undergoing IS therapy; following bone marrow transplantation, the risk of reactivation may exceed 75% [79]. The risk of HBV recurrence during IS therapy is generally lower, but may exceed 10% in some cases, particularly with treatments leading to B-cell depletion [80,81].
During prophylaxis, ALT and HBV DNA are monitored every 3–6 months and for at least 12 months after the end of NA prophylaxis. A significant proportion of reactivations occur only after discontinuation of NA.
The main virological manifestation of recurrence is the reappearance of HBsAg in serum (seroreversion), consistently accompanied by an exacerbation of hepatitis (flare). In contrast, the reappearance of positive serum HBV DNA is accompanied by seroreversion and exacerbation of hepatitis in only about half of cases. In the event of HBV infection recurrence, characterized by detectable serum HBV DNA or HBsAg seroconversion, it is essential to immediately initiate NA treatment (ETV, TDF, or TAF), preferably even before an increase in ALT activity. HBsAg seroconversion can lead to severe, even fulminant, acute hepatitis [3].
Prevention of HBV Recurrence After Liver and Other Organ Transplants
The source of HBV infection in cases of recurrence after liver transplantation (LT) can be either the recipient, if the graft is from an anti-HBc-negative donor and there is insufficient prophylaxis against recurrence, or the donor, if the graft is from an anti-HBc-positive donor and contains cccDNA. Even in the case of infection from an anti -HBc-positive donor, the term HBV recurrence is used because HBV infection is reactivated. Hepatitis resulting from HBV recurrence is usually severe and often leads to organ failure (if left untreated). A particularly rapidly progressive form is referred to as fibrosing cholestatic hepatitis B [82].
HBV recurrence is characterized by HBsAg positivity and/or HBV DNA positivity and can take the following forms:
- positivity for both HBsAg and HBV DNA – the most common;
- HBsAg negativity and HBV DNA positivity – a rare situation arising from the development of “escape” mutations in the HBsAg gene [83].
- The standard recommended prophylaxis to prevent recurrence of HBV infection after liver transplantation is a combination of ETV, TDV, or TAF and hyperimmune immunoglobulin against HBV (HBIG).
- TAF or ETV is preferred because treatment with calcineurin inhibitors and corticosteroids carries a higher risk of renal insufficiency and reduced bone density, which TDF could exacerbate.
- HBIG administration begins during the pre-liver transplant phase, and post-transplant dosing is guided by anti- -HBs—the target concentration is ≥ 50–100 IU/L.
- HBIG administration may be discontinued after TJ if the patient has good adherence to NA therapy and the risk of recurrence is low.
- Recommendations for discontinuing HBIG vary. Most agree that if long-term HBsAg negativity, anti-HBs positivity, and HBV DNA negativity are achieved through combined prophylaxis, HBIG prophylaxis can be discontinued and NA continued.
- Discontinuation of HBIG is not recommended in cases of high viremia at the time of LT (> 100,000 IU/ml) or evidence of HCC prior to LT.
- In the event of seroconversion after LT, HBIG administration is discontinued and NA administration is continued [3].
HBV prophylaxis without HBIG (HBIG-free prophylaxis), i.e., “only” by administering effective NA, may be considered if the risk of HBV recurrence is low—HBV DNA is negative at the time of TJ and there is no HDV or HIV coinfection, nor HCC. A prerequisite for the application of this strategy is effective NA treatment of the recipient prior to TJ. With HBIG-free prophylaxis, HBsAg positivity occurs more frequently after TJ than with combined prophylaxis, but it is unclear whether this represents incomplete elimination of HBsAg or true recurrence of HBV infection. HBsAg positivity is often transient, and its clinical significance appears to be minimal [84].
Preventing HBV reinfection after HCT is crucial for the final outcome of HDV infection, as HDV infection during HCT is highly aggressive and there is insufficient robust data on the use of BLV for this indication. Therefore, combined NA and HBIG prophylaxis is necessary for this indication. The gold standard is lifelong administration of NA and HBIG; with a minimum duration of 24 months post-LT [67].
Most patients with HIV infection are HIV RNA- and HBV DNA-negative at the time of LT, so the recommendations for prophylaxis are essentially the same as for HBV monoinfection. It is important that ART include TAF or TDF. There is currently insufficient data to support discontinuation of HBIG or an HBIG-free regimen immediately after LT [85,86].
Patients with chronic HBV infection and HCC have a higher risk of HBV recurrence after LT (2–35%) than those without HCC (1.9–9.7%). The risk is particularly elevated in advanced HCC [87]. HBV recurrence after LT is significantly associated with HCC recurrence in the liver and/or extrahepatic sites. HCC recurrence after LT has been observed in 10–15% of HBsAg-positive patients. HBV DNA or HBsAg positivity in the liver recipient therefore raises a reasonable suspicion of HCC recurrence [88]. Currently, discontinuation of HBIG prophylaxis or an HBIG-free regimen immediately after LT is not recommended in these patients.
Table 15 lists all measures that must be taken in the case of organ transplantation from an anti-HBc-positive donor to a recipient who is HBsAg-negative [3].

Liver transplantation from an HBsAg-positive donor is an exceptional solution to the shortage of suitable donors—such livers are primarily transplanted to HBsAg-positive recipients [89]. All liver recipients must receive prophylactic ETV, TDF, or TAF; HBIG prophylaxis is not necessary because the donor liver is already infected with HBV and reinfection cannot be prevented. A patient with chronic hepatitis D should not receive a liver from an HBsAg-positive donor.
Transplantation of other organs from an HBsAg-positive donor is an exceptional solution to the shortage of suitable donors—such organs are primarily transplanted to HBsAg-positive recipients; in the case of an HBsAg-negative recipient, active and passive immunization against HBV prior to transplantation is recommended. Recipients of other organs must receive prophylactic ETV, TDF, or TAF and HBIG (unless the recipient has an anti-HBs level >100 IU/ml) [90]. In the case of bone marrow or solid organ transplantation from a living donor, the donor should be treated with effective NAs as soon as possible prior to transplantation [91].
HBV Vaccination
HBV vaccines were initially derived from plasma (early 1980s), then replaced by a series of second-generation vaccines produced by genetically engineered mammalian or yeast cells. Their introduction into clinical practice has drastically reduced the transmission of HBV infection (including vertical, nosocomial, and occupational transmission) and the incidence of both acute and chronic forms of HBV infection [92]. Conventional second-generation recombinant vaccines containing the S antigen induce robust immunogenicity in young, healthy individuals – the vaccination regimen consists of three doses. The efficacy of these vaccines decreases in older individuals and those with comorbidities – the protective level of anti-HBs antibodies (≥ 10 IU/ml) is achieved after 3 vaccine doses in < 75% of those vaccinated. Obesity, smoking, male gender, immunosuppression, and chronic diseases (liver cirrhosis, chronic kidney disease, and diabetes mellitus) are associated with reduced vaccine efficacy [93,94] . Pregnant women may be vaccinated against HBV, although the indication must be carefully considered, as with all vaccinations during pregnancy [95].
Third-generation vaccines are more effective, particularly in subgroups of vaccinated individuals who respond suboptimally to conventional vaccination with second-generation vaccines [96].
Universal vaccination in childhood is recommended as early as possible, at the latest before puberty. In the Czech Republic, universal vaccination of children against HBV has been carried out since 2001—now starting at 9 weeks of age.
- Properly vaccinated individuals who are healthy and under 40 years of age do not need to be tested for anti-HBs levels; a protective anti-HBs titer (> 10 IU/L) is achieved in ≥ 95% of children and ≥ 90% of healthy adults.
- The anti-HBs titer may drop below 10 IU/L within 4–10 years in approximately 10–50% of healthy individuals; however, these individuals remain protected against HBV due to robust immune memory that persists even after the anti-HBs titer declines.
- Revaccination of individuals who have lost their protective anti-HBs level leads to a significant rise in anti-HBs titer within 3–7 days.
- This “anamnestic” immune response is detectable in more than 70% of vaccinated individuals up to 35 years after completion of the primary vaccination series.
- According to the 2025 EASL Recommendations, a value of ≥ 100 IU/ml should be considered a protective anti-HBs titer, particularly in individuals at increased risk of severe infection, such as immunocompromised individuals [3].
The combination of active immunization (vaccination) and passive immunization is of crucial importance in preventing mother-to-newborn transmission, which otherwise occurs primarily during childbirth and breastfeeding if the newborn has not been properly immunized.
- Newborns of HBsAg-positive mothers should receive the first dose of the vaccine as soon as possible after birth (no later than 12 hours of age, but preferably within the first hour of life) in combination with passive immunization using hyperimmune immunoglobulin against HBV (HBIG).
- The combination of vaccination and passive immunization results in seroprotection in 95% of healthy, full-term newborns.
- Failure of this approach is usually associated with HBeAg positivity or high HBV DNA levels in the mother.
- Newborns with a birth weight < 2,000 g have a reduced response to vaccination, and it is recommended that they receive 4 doses of the vaccine.
- Post-vaccination testing (anti-HBs, HBsAg) is usually performed between 9 and 12 months of age—if anti-HBs < 10 IU/L, a fourth dose of the vaccine is administered and testing is performed 1–2 months later; if anti-HBs are still < 10 IU/L, a fifth and sixth dose of the vaccine are administered, and testing is performed 1–2 months after the sixth dose [3].
Vaccination of immunosuppressed or immunodeficient individuals should be administered as a double dose of the standard second-generation vaccine or a third-generation vaccine. Patients undergoing hemodialysis respond better to a double dose of the second-generation vaccine or to the third-generation vaccine [97]. This also applies to chronic alcoholics [98] . Immunocompetent HIV-positive individuals are vaccinated in the same manner as healthy individuals; however, double doses of the conventional vaccine are administered when CD4+ counts are low [99]. For the vaccination of patients with cirrhosis, a double dose of the conventional vaccine is recommended, but even 6 doses of the vaccine may result in a suboptimal response [100]. A third-generation vaccine may be more effective for this indication [101].
The seroprotective concentration of anti-HBs has been established as ≥ 10 IU/L, but in high-risk individuals, such as those who are immunosuppressed, an anti-HBs concentration of ≥ 100 IU/L measured 1–2 months after the last vaccine dose is considered the optimal effect of vaccination—this indicates long-term, possibly lifelong protection against HBV. Individuals with an anti- -HBs ≥ 100 IU/L measured 1–2 months after the last vaccine dose do not need further testing, with the exception of immunocompromised individuals, who should be tested every 1–10 years depending on the level of risk—if anti -HBs < 100 IU/L, a booster dose of the vaccine is warranted. At-risk individuals with anti-HBs levels between 10 and 100 IU/L 1–2 months after the last vaccine dose should receive a fourth dose of the vaccine, followed by a check of anti-HBs levels after 1–2 months. Individuals with an anti-HBs titer < 10 IU/L measured 1–2 months after the last vaccine dose are recommended to undergo
complete revaccination with 3 vaccine doses, possibly as a double single dose, followed by anti-HBs monitoring 1–2 months after completion of the second vaccination. Prior to revaccination, it is advisable to check for ongoing HBV infection (HBsAg, anti-HBc) [3]. Intradermal vaccination of non-responders, although it appears to be immunologically advantageous, has not yet demonstrated superior efficacy in immunocompromised individuals [102].
Management of patients with hepatitis B virus infection
In the Czech Republic, the law mandates the mandatory isolation of patients with acute hepatitis in infectious disease clinics or wards. Furthermore, patients are monitored for at least 12 months in liver clinics at these facilities. If the infection becomes chronic, long-term, or possibly lifelong follow-up is required in liver clinics at infectious disease, gastroenterology, or internal medicine departments. In the event of HCC development, follow-up occurs in gastroenterology and oncology departments. Only facilities accredited by the Czech Society of Hepatology and the Society of Infectious Diseases of the J. E. Purkyně Czech Medical Society are authorized to provide antiviral treatment for chronic hepatitis B.
Korespondenční autor
prof. MUDr. Petr Husa, CSc.
Klinika infekčních chorob
LF MU a FN Brno
Jihlavská 20
625 00 Brno
husa.petr@fnbrno.cz
Literatura
1. Husa P, Šperl J, Urbánek P et al. Doporučený postup gnostiky a léčby chronické hepatitidy B. Gastroent Hepatol 2014; 68(6): 514–526. doi: 10.14735/amgh2014514.
2. Husa P, Šperl J, Urbánek P et al. Diagnosis and therapy of chronic hepatitis B: Czech national guidelines. Klin Mikrobiol Infect Lek 2014; 20(4): 121–132.
3. European Association for the Study of the Liver. EASL Clinical Practice Guidelines on the management of hepatitis B virus infection. J Hepatol 2025; 83(2): 502–583. doi: 10.1016/j.jhep.2025.03.018
4. WHO. Global hepatitis report 2024: action for accession low- and middle-income countries. 2024 [online]. Dostupné z: https: //www.who.int/publications/i/item/9789240091672.
5. Polaris Observatory Collaborators. Global prevalence, cascade of care, and prophylaxis coverage of hepatitis B in 2022: a modelling study. Lancet Gastroenterol Hepatol 2023; 8(10): 879–907. doi: 10.1016/S2468-1253(23)00197-8.
6. Němeček V, Částková J, Fritz P et al. The 2001 serological survey in the Czech Republic – viral hepatitis. Cent Eur J Public Health 2003; 47(Suppl 1): S54–S61.
7. Bílková Fránková H, Kloudová A, Zelená H et al. Víceúčelový sérologický přehled (spalničky, příušnice, pertusse, virová hepatitida B) SP 2013, ČR: závěrečná zpráva, příloha č. 1. Zprávy CEM (SZÚ Praha) 2014; 23: 1–152. 2015 [online]. Dostupné z: http: //www.zuusti.cz/wp-content/uploads/2015/04/SP-2013-vnitřek-do-TISKU.pdf.
8. ISIN. Epidemiologická databáze Státního zdravotního ústavu Praha. 2025 [online]. Dostupné z: http: //www.szu.cz/publikace/data/infekce-v-cr.
9. Husa P, Urbánek P. Virová hepatitida B. In: Urbánek P, Brůha R (eds). Hepatologie. Praha: Grada Publishing: 2022: 343–355.
10. Huang DQ, Tran A, Yeh M-L et al. Antiviral therapy substantially reduces HCC risk in patients with chronic hepatitis B infection in the indeterminate phase. Hepatology 2023; 78(5): 1558–1568. doi: 10.1097/HEP.0000 000000000459.
11. Lai JC, Wong GL, Tse YK et al. Histological severity, clinical outcomes and impact of antiviral treatment in indeterminate phase of chronic hepatitis B: a systematic review and meta-analysis. J Hepatol 2025; 82(6): 992–1003. doi: 10.1016/j.jhep.2024.11.018.
12. Yapali S, Talaat N, Fontana RJ et al. Outcomes of patients with chronic hepatitis B who do not meet criteria for antiviral treatment at presentation. Clin Gastroenterol Hepatol 2015; 13(1): 193.e1–201.e1. doi: 10.1016/j.cgh.2014.07.019.
13. Pronier C, Candotti D, Boizeau L et al. The contribution of more sensitive hepatitis B surface antigen assays to detecting and monitoring hepatitis B infection. J Clin Virol 2020; 129: 104507. doi: 10.1016/j.jcv.2020.104507.
14. Fu MX, Simmonds P, Andreani J et al. Ultrasensitive PCR system for HBV DNA detection: risk stratification for occult hepatitis B virus infection in English blood donors. J Med Virol 2023; 95(10): e29144. doi: 10.1002/jmv.29144.
15. Duarte-Rojo A, Taouli B, Leung DH et al. Imaging-based noninvasive liver disease assessment for staging liver fibrosis in chronic liver disease: a systematic review supporting the AASLD Practice Guideline. Hepatology 2025; 81(2): 725–748. doi: 10.1097/HEP.0000000000000852.
16. de Franchis R, Baveno VI Faculty. Expanding consensus in portal hypertension: report of the Baveno VI Consensus Workshop: stratifying risk and individualizing care for portal hypertension. J Hepatol 2015; 63(3): 743–752. doi: 10.1016/j.jhep.2015.05.022.
17. Papatheodoridi M, Hiriart JB, Lupsor-Platon M et al. Refining the Baveno VI elastography criteria for the definition of compensated advanced chronic liver disease. J Hepatol 2021; 74(5): 1109–1116. doi: 10.1016/j.jhep.2020.11.050.
18. Cornberg M, Wong VW-S, Locarnini S et al. The role of quantitative hepatitis B surface antigen revisited. J Hepatol 2017; 66(2): 398–411. doi: 10.1016/j.jhep.2016.08.009.
19. Kramvis A, Chang KM, Dandri M et al. A roadmap for serum biomarkers for hepatitis B virus: current status and future outlook. Nat Rev Gastroenterol Hepatol 2022; 19(11): 727–745. doi: 10.1038/s41575-022-00649-z.
20. Yoshida K, Desbiolles A, Feldman SF et al. Hepatitis B core-related antigen to indicate high viral load: systematic review and meta-analysis of 10,397 individual participants. Clin Gastroenterol Hepatol 2021; 19(1): 46–60.e8. doi: 10.1016/j.cgh.2020.04.045.
21. Shimakawa Y, Ndow G, Kaneko A et al. Rapid point-of-care test for hepatitis B core-related antigen to gnosehigh viral load in resource-limited settings. Clin Gastroenterol Hepatol 2023; 21(7): 1943.e2–1946.e2. doi: 10.1016/ j.cgh.2022.05.026.
22. Chen CJ, Yang HI, Su J et al. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. J Am Med Assoc 2006; 295(1): 65–73. doi: 10.1001/jama.295.1.65.
23. Iloeje UH, Yang HI, Su J et al. Predicting cirrhosis risk based on the level of circulating hepatitis B viral load. Gastroenterology 2006; 130(3): 678–686. doi: 10.1053/j.gastro.2005.11.016.
24. Chen X, Wu F, Liu Y et al. The contribution of serum hepatitis B virus load in the carcinogenesis and prognosis of hepatocellular carcinoma: evidence from two meta-analyses. Oncotarget 2016; 7(31): 49299–49309. doi: 10.18632/oncotarget.10335.
25. Chen CJ, Yang HI, Iloeje UH et al. Hepatitis B virus DNA levels and outcomes in chronic hepatitis B. Hepatology 2009; 49(Suppl 5): S72–S84. doi: 10.1002/hep.22884.
26. Gerlich WH. Reduction of infectivity in chronic hepatitis B virus carriers among healthcare providers and pregnant women by antiviral therapy. Intervirology 2014; 57(3–4): 202–211. doi: 10.1159/000360949.
27. Incident Investigation Teams and Others. Transmission of hepatitis B to patients from four infected surgeons without hepatitis B e antigen. N Engl J Med 1997; 336(3): 178–184. doi: 10.1056/NEJM199701163360304.
28. Yoo JJ, Jung EA, Kim SG et al. Risk of dyslipidaemia in people living with HIV who are taking tenofovir alafenamide: a systematic review and meta-analysis. J Int AIDS Soc 2024; 27(9): e26358. doi: 10.1002/jia2.26358.
29. Lin S, Huang W, Liao Z et al. Comparison of lipid profile alterations in chronic hepatitis b patients receiving tenofovir alafenamideor tenofovir disoproxil fumarate. Sci Rep 2024; 14(1): 27369. doi: 10.1038/s41598-024-78656-0.
30. Hwang EG, Jung EA, Yoo JJ et al. Risk of dyslipidemia in chronic hepatitis B patients taking tenofovir alafenamide: a systematic review and meta-analysis. Hepatol Int 2023; 17(4): 860–869. doi: 10.1007/s12072-023-10528-7.
31. Mallon PWG, Brunet L, Fusco JS et al. Lipid changes after switch from TDF to TAF in the OPERA cohort: LDL cholesterol and triglycerides. Open Forum Infect Dis 2022; 9(1): ofab621. doi: 10.1093/ofid/ofab621.
32. Kanters S, Renaud F, Rangaraj A et al. Evidence synthesis evaluating body weight gain among people treating HIV with antiretroviral therapy-a systematic literature review and network meta-analysis. EClinicalMedicine 2022; 48: 101412. doi: 10.1016/j.eclinm.2022. 101412.
33. Zoutendijk R, Reijnders JG, Zoulim F et al. Virological response to entecavir is associated with a better clinical outcome in chronic hepatitis B patients with cirrhosis. Gut 2013; 62(5): 760–765. doi: 10.1136/gutjnl-2012-302024.
34. Zoulim F, Locarnini S. Hepatitis B virus resistance to nucleos (t) ide analogues. Gastroenterology 2009; 137(5): 1593–1608. doi: 10.1053/j.gastro.2009.08.063.
35. Sherman M, Yurdaydin C, Sollano J et al. Entecavir for treatment of lamivudine-refractory, HBeAg-positive chronic hepatitis B. Gastroenterology 2006; 130(7): 2039–2049. doi: 10.1053/j.gastro.2006.04.007.
36. Kitrinos KM, Corsa A, Liu Y et al. No detectable resistance to tenofovir disoproxil fumarate after 6 years of therapy in patients with chronic hepatitis B. Hepatology 2014; 59(2): 434–442. doi: 10.1002/hep.26686.
37. Chan HL, Buti M, Lim YS et al. Long-term treatment with tenofovir alafenamide for chronic hepatitis B results in high rates of viral suppression and favorable renal and bone safety. Am J Gastroenterol 2024; 119(3): 486–496. doi: 10.14309/ajg.0000000000002468.
38. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: management of hepatocellular carcinoma. J Hepatol 2018; 69(1): 182–236. doi: 10.1016/j.jhep.2018.03.019.
39. European Association for the Study of the Liver. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J Hepatol 2017; 67(2): 370–398. doi: 10.1016/j.jhep.2017.03.021.
40. Wiegand J, Wedemeyer H, Franke A et al. Treatment of severe, nonfulminant acute hepatitis B with lamivudine vs placebo: a prospective randomized double-blinded multicentre trial. J Viral Hepat 2014; 21(10): 744–750. doi: 10.1111/jvh.12210.
41. Mantzoukis K, Rodríguez-Perálvarez M, Buzzetti E et al. Pharmacological interventions for acute hepatitis B infection: an attempted network meta-analysis. Cochrane Database Syst Rev 2017; 3(3): CD011645. doi: 10.1002/14651858.CD011645.pub2.
42. Yu JW, Sun LJ, Zhao YH et al. The study of efficacy of lamivudine in patients with severe acute hepatitis B. Dig Dis Sci 2010; 55(3): 775–783. doi: 10.1007/s10620-009-1060-5.
43. Brown RS, McMahon BJ, Lok ASF et al. Antiviral therapy in chronic hepatitis B viral infection during pregnancy: a systematic review and meta-analysis. Hepatology 2016; 63(1): 319–333. doi: 10.1002/hep.28302.
44. Li W, Jia L, Zhao X et al. Efficacy and safety of tenofovir in preventing mother-to-infant transmission of hepatitis B virus: a meta-analysis based on 6 studies from China and 3 studies from other countries. BMC Gastroenterol 2018; 18(1): 121. doi: 10.1186/s12876-018-0847-2.
45. Shang J, Wen Q, Wang CC et al. Safety and efficacy of telbivudine for chronic hepatitis B during the entire pregnancy: long-term follow-up. J Viral Hepat 2017; 24(Suppl 1): 43–48. doi: 10.1111/jvh.12785.
46. Zeng QL, Yu ZJ, Ji F et al. Tenofovir alafenamide to prevent perinatal hepatitis B transmission: a multicenter, prospective, observational study. Clin Infect Dis 2021; 73(9): e3324–e3332. doi: 10.1093/cid/ciaa1939.
47. Ding Y, Cao L, Zhu L et al. Efficacy and safety of tenofovir alafenamide fumarate for preventing mother-to-child transmission of hepatitis B virus: a national cohort study. Aliment Pharmacol Ther 2020; 52(8): 1377–1386. doi: 10.1111/apt.16043.
48. Chen R, Zou J, Long L et al. Safety and efficacy of tenofovir alafenamide fumarate in early-middle pregnancy for mothers with chronic hepatitis B. Front Med (Lausanne) 2021; 8: 796901. doi: 10.3389/fmed.2021.796901.
49. Pan CQ, Zhu L, Yu AS et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate for preventing vertical transmission in chronic hepatitis B mothers: a systematic review and meta-analysis. Clin Infect Dis 2024; 79(4): 953–964. doi: 10.1093/cid/ciae288.ciae288.
50. Wen WH, Chang MH, Zhao LL et al. Motherto-infant transmission of hepatitis B virus infection: significance of maternal viral load and strategies for intervention. J Hepatol 2013; 59(1): 24–30. doi: 10.1016/j.jhep.2013.02.015.
51. Pan CQ, Duan ZP, Bhamidimarri KR et al. Analgorithm for risk assessment and intervention of mother to child transmission of hepatitis B virus. Clin Gastroenterol Hepatol 2012; 10(5): 452–459. doi: 10.1016/j.cgh.2011.10.041.
52. You H, Wang F, Li T et al. Guidelines for the prevention and treatment of chronic hepatitis B (version 2022). J Clin Transl Hepatol 2023; 11(6): 1425–1442. doi: 10.14218/JCTH.2023.00320.
53. Ehrhardt S, Xie C, Guo N et al. Breastfeeding while taking lamivudine or tenofovir disoproxil fumarate: a review of the evidence. Clin Infect Dis 2015; 60(2): 275–278. doi: 10.1093/cid/ciu798.
54. Hu X, Wang L, Xu F. Guides concerning tenofovir exposure via breastfeeding: a comparison of drug dosages by developmental stage. Int J Infect Dis 2019; 87: 8–12. doi: 10.1016/j.ijid.2019.07.023.
55. Li S, Jin J, Jiang Y et al. Low levels of tenofovir in breast milk support breastfeeding in HBV-infected mothers treated with tenofovir disoproxil fumarate. Int J Antimicrob Agents 2023; 61(3): 106726. doi: 10.1016/j.ijantimicag.2023.106726.
56. Li B, Liu Z, Liu X et al. Efficacy and safety of tenofovir disoproxil fumarate and tenofovir alafenamide fumarate in preventing HBV vertical transmission of high maternal viral load. Hepatol Int 2021; 15(5): 1103–1108. doi: 10.1007/s12072-021-10235-1.
57. Pan YC, Jia ZF, Wang YQ et al. The role of caesarean section and nonbreastfeeding in preventing mother-to-childtransmission of hepatitis B virus in HBsAg-and HBeAg-positive mothers: results from a prospective cohort study and a meta-analysis. J Viral Hepat 2020; 27(10): 1032–1043. doi: 10.1111/jvh.13314.
58. Levy MT, Terrault NA. Caesarean section or non-breastfeeding for prevention of MTCT-beware of sending the wrong message. J Viral Hepat 2021; 28(3): 575–576. doi: 10.1111/jvh.13455.
59. Yang M, Qin Q, Fang Q et al. Cesarean section to prevent mother-to-child transmission of hepatitis B virus in China: a meta-analysis. BMC Pregnancy Childbirth 2017; 17(1): 303. doi: 10.1186/s12884-017-1487-1.
60. Chen HL, Cai JY, Song YP et al. Vaginal delivery and HBV mother to child transmission risk after immunoprophylaxis: a systematic review and a meta-analysis. Midwifery 2019; 74: 116–125. doi: 10.1016/j.midw.2019.03.024.
61. Pan CQ, Zou HB, Chen Y et al. Cesarean section reduces perinatal transmission of hepatitis B virus infection from hepatitis B surface antigen-positive women to their infants. Clin Gastroenterol Hepatol 2013; 11(10): 1349–1355. doi: 10.1016/j.cgh.2013.04.026.
62. Singal AK, Salameh H, Kuo YF et al. Meta-analysis: the impact of oral anti-viral agents on the incidence of hepatocellular carcinoma inchronic hepatitis B. Aliment Pharmacol Ther 2013; 38(2): 98–106. doi: 10.1111/apt.12344.
63. Shim JH, Lee HC, Kim KM et al. Efficacy of entecavir in treatment-naïve patients with hepatitis B virus-related decompensated cirrhosis. J Hepatol 2010; 52(2): 176–182. doi: 10.1016/j.jhep.2009.11.007.
64. Wang N, He S, Zheng Y et al. Efficacy and safety of tenofovir disoproxil fumarate versus entecavir in the treatment of acute-on-chronic liver failure with hepatitis B: a systematic review and meta-analysis. BMC Gastroenterol 2023; 23(1): 388. doi: 10.1186/s12876-023-03024-7.
65. Li J, Hu C, Chen Y et al. Short-term and long-term safety and efficacy of tenofovir alafenamide, tenofovir disoproxil fumarate and entecavir treatment of acute-on-chronic liver failure associated with hepatitis B. BMC Infect Dis 2021; 21(1): 567. doi: 10.1186/s12879-021-06237-x.
66. Peng W, Gu H, Cheng D et al. Tenofovir alafenamide versus entecavir for treating hepatitis B virus-related acute-onchronic liver failure: real-world study. Front Microbiol 2023; 14: 1185492. doi: 10.3389/fmicb.2023.1185492.
67. European Association for the Study of the Liver. EASL Clinical Practice Guidelines on hepatitis delta virus. J Hepatol 2023; 79(2): 433–460. doi: 10.1016/j.jhep.2023.05.00.
68. Husa P. Šperl J, Urbánek P et al. Doporučený postup gnostiky a léčby chronické hepatitidy D. Datum vydání doporučení. 2023. Vnitr Lek 2023; 69(8): 525–532. doi: 10.36290/vnl.2023.104.
69. Husa P, Šperl J, Urbánek P et al. Doporučený postup gnostiky a léčby chronické hepatitidy D. Klin Mikrobiol Inf Lek 2024; 29(Suppl 3): 1–9.
70. Degasperi E, Anolli MP, Uceda Renteria SC et al. Bulevirtide monotherapy for 48 weeks in patients with HDV-related compensated cirrhosis and clinically significant portal hypertension. J Hepatol 2022; 77(6): 1525–1531. doi: 10.1016/j.jhep.2022.07.016.
71. Dietz-Fricke C, Tacke F, Zöllner C et al. Treating hepatitis D with bulevirtide – real-world experience from 114 patients. JHEP Rep 2023; 5(4): 100686. doi: 10.1016/j.jhepr.2023.100686.
72. Leumi S, Bigna JJ, Amougou MA et al. Global burden of hepatitis B infection in people living with human immunodeficiency virus: a systematic review and meta-analysis. Clin Infect Dis 2020; 71(11): 2799–2806. doi: 10.1093/cid/ciz1170.
73. Kim HN, Newcomb CW, Carbonari DM et al. Risk of HCC with hepatitis B viremia among HIV/HBV-coinfected persons in North America. Hepatology 2021; 74(3): 1190–1202. doi: 10.1002/hep.31839.
74. Donato F, Boffetta P, Puoti M. A meta-analysis of epidemiological studies on the combined effect of hepatitis B and C virus infections in causing hepatocellular carcinoma. Int J Cancer 1998; 75(3): 347–354. doi: 10.1002/(SICI)1097-0215(19980130)75: 3< 347::aid-ijc4>3.0.co;2-2.
75. Shi J, Zhu L, Liu S et al. A meta-analysis of case-control studies on the combined effect of hepatitis B and C virus infections in causing hepatocellular carcinoma in China. Br J Cancer 2005; 92(3): 607–612. doi: 10.1038/sj.bjc.6602333.
76. Mücke MM, Backus LI, Mücke VT et al. Hepatitis B virus reactivation during direct-acting antiviral therapy for hepatitis C: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol 2018; 3(3): 172–180. doi: 10.1016/S2468-1253(18)30002-5.
77. Serper M, Forde KA, Kaplan DE. Rare clinically significant hepatic events and hepatitis B reactivation occur more frequently following rather than during direct-acting antiviral therapy for chronic hepatitis C: data from a national US cohort. J Viral Hepat 2018; 25(2): 187–197. doi: 10.1111/jvh.12784.
78. Jaroszewicz J, Pawłowska M, Simon K et al. Low risk of HBV reactivation in a large European cohort of HCV/HBV coinfected patients treated with DAA. Expert Rev Anti Infect Ther 2020; 18(10): 1045–1054. doi: 10.1080/14787210.2020.1782189.
79. Papatheodoridis GV, Lekakis V, Voulgaris T et al. Hepatitis B virus reactivation associated with new classes of immunosuppressants and immunomodulators: a systematic review, meta-analysis, and expert opinion. J Hepatol 2022; 77(6): 1670–1689. doi: 10.1016/j.jhep.2022.07.003.
80. Cholongitas E, Haidich AB, Apostolidou-Kiouti F et al. Hepatitis B virus reactivation in HBsAg-negative, anti-HBc-positive patients receiving immunosuppressive therapy: a systematic review. Ann Gastroenterol 2018; 31(4): 480–490. doi: 10.20524/aog.2018.0266.
81. Mozessohn L, Chan KK, Feld JJ et al. Hepatitis B reactivation in HBsAg-negative/HBcAb-positive patients receiving rituximab for lymphoma: a meta-analysis. J Viral Hepat 2015; 22(10): 842–849. doi: 10.1111/jvh.12402.
82. Samuel D, Muller R, Alexander G et al. Liver transplantation in European patients with the hepatitis B surface antigen. N Engl J Med 1993; 329(25): 1842–1847. doi: 10.1056/NEJM 199312163292503.
83. Duvoux C, Belli LS, Fung J et al. 2020 position statement and recommendations of the European Liver and Intestine Transplantation Association (ELITA): management of hepatitis B virusrelated infection before and after liver transplantation. Aliment Pharmacol Ther 2021; 54(5): 583–605. doi: 10.1111/apt.16374.
84. Fung J, Wong T, Chok K et al. Long-term outcomes of entecavir monotherapy for chronic hepatitis B after liver transplantation: results up to 8 years. Hepatology 2017; 66(4): 1036–1044. doi: 10.1002/hep.29191.
85. Agüero F, Forner A, Manzardo C et al. Human immunodeficiency virus infection does not worsen prognosis of liver transplantation for hepatocellular carcinoma. Hepatology 2016; 63(2): 488–498. doi: 10.1002/hep.28321.
86. Jacob JS, Shaikh A, Goli K et al. Improved survival after liver transplantation for patients with human immunodeficiency virus (HIV) and HIV/hepatitis C virus coinfection in the integrase strand transfer inhibitor and direct-acting antiviral eras. Clin Infect Dis 2023; 76(4): 592–599. doi: 10.1093/cid/ciac821.
87. Faria LC, Gigou M, Roque-Afonso AM et al. Hepatocellular carcinoma is associated with an increased risk of hepatitis B virus recurrence after liver transplantation. Gastroenterology 2008; 134(7): 1890–1899. doi: 10.1053/ j.gastro.2008.02.064.
88. Saab S, Yeganeh M, Nguyen K et al. Recurrence of hepatocellular carcinoma and hepatitis B reinfection in hepatitis B surface antigen-positive patients after liver transplantation. Liver Transpl 2009; 15(11): 1525–1534. doi: 10.1002/lt.21882.
89. European Association for the Study of the Liver. EASL Clinical Practice Guidelines on liver transplantation. J Hepatol 2024; 81(6): 1040–1086. doi: 10.1016/j.jhep.2024.07.032.
90. Chancharoenthana W, Townamchai N, Pongpirul K et al. The outcomes of kidney transplantation in hepatitis B surface antigen (HBsAg) -negative recipients receiving graft from HBsAg-positive donors: a retrospective, propensity score-matched study. Am J Transplant 2014; 14(12): 2814–2820. doi: 10.1111/ajt.12921.
91. Hui CK, Lie A, Au WY et al. Effectiveness of prophylactic anti-HBV therapy in allogeneic hematopoietic stem cell transplantation with HBsAg positive donors. Am J Transplant 2005; 5(6): 1437–1445. doi: 10.1111/j.1600-6143. 2005.00887.x.
92. Wong GL, Hui VW, Yip TC et al. Universal HBV vaccination dramatically reduces the prevalence of HBV infection and incidence of hepatocellular carcinoma. Aliment Pharmacol Ther 2022; 56(5): 869–877. doi: 10.1111/apt.17120.
93. Fan W, Chen XF, Shen C et al. Hepatitis B vaccine response in obesity: a meta-analysis. Vaccine 2016; 34(40): 4835–4841. doi: 10.1016/j.vaccine.2016.08.027.
94. Aggeletopoulou I, Davoulou P, Konstantakis C et al. Response to hepatitis B vaccination in patients with livercirrhosis. Rev Med Virol 2017; 27(6). doi: 10.1002/rmv.1942.
95. Sheffield JS, Hickman A, Tang J et al. Efficacy of an accelerated hepatitis b vaccination program during pregnancy. Obstet Gynecol 2011; 117(5): 1130–1135. doi: 10.1097/AOG. 0b013e3182148efe.
96. A two-dose hepatitis B vaccine for adults (Heplisav-B). JAMA 2018; 319(8): 822–823. doi: 10.1001/jama.2018.1097.
97. Awad AM, Ntoso A, Connaire JJ et al. An open-label, single-arm study evaluating the immunogenicity and safety of the hepatitis B vaccine HepB-CpG (HEPLISAV-B®) in adults receiving hemolysis. Vaccine 2021; 39(25): 3346–3352. doi: 10.1016/j.vaccine.2021.05.003.
98. Rosman AS, Basu P, Galvin K et al. Efficacy of a high and accelerated dose of hepatitis B vaccine in alcoholic patients: a randomized clinical trial. Am J Med 1997; 103(3): 217–222. doi: 10.1016/s0002-9343(97)00132-0.
99. Gupta A, Fine SM, Vail RM et al. Prevention and management of hepatitis B virus infection in adults with HIV. Baltimore (MD): Johns Hopkins University 2022.
100. Rodríguez-Tajes S, Pocurull A, Lens S et al. Efficacy of an accelerated double-dose hepatitis B vaccine regimen in patients with cirrhosis. J Viral Hepat 2021; 28(7): 1019–1024. doi: 10.1111/jvh.13509.
101. Amjad W, Alukal J, Zhang T et al. Two-dose hepatitis B vaccine (Heplisav-B) results in better seroconversion than three-dose vaccine (Engerix-B) in chronic liver disease. Dig Dis Sci 2021; 66(6): 2101–2106. doi: 10.1007/s10620-020-06437-6.
102. David MC, Ha SH, Paynter S et al. A systematic review and metaanalysis of management options for adults who respond poorly to hepatitis B vaccination. Vaccine 2015; 33(48): 6564–6569. doi: 10.1016/j.vaccine.2015.09.051.