
Klíčová slova
výzkum a vývoj
indikace
klinické studie
biosimilars
Abstrakt
Chimérickou monoklonální protilátku proti tumor nekrotizujícímu faktoru (TNF), pro kterou dnes používáme generický název infliximab, vyvinuli J. Le a J. Vilček na New York University School of Medicine ve spolupráci s firmou Centocor (později Janssen Biotech) pod názvem cA2. V roce 1992 ji M. Feldmann a R. Maini použili v první proof-of-concept studii u revmatoidní artritidy k průkazu hypotézy, že TNF stojí na vrcholu zánětlivé kaskády autoimunitních zánětů. V roce 1998 ji americká Food and Drug Administration (FDA) schválila pod výrobním názvem Remicade pro léčbu Crohnovy nemoci. Mezi lety 1999 a 2006 byla registrována pro léčbu řady dalších autoimunitních onemocnění. Jako jeho první biologicky podobný léčivý přípravek (biosimilar) byl v roce 2014 registrován přípravek s označením CT-P13 vyvinutý firmou Celltrion. Infliximab zásadním způsobem posunul možnosti farmakoterapie řady autoimunitních onemocnění, ale přispěl i ke studiu jejich patofyziologie a k pokroku biologické léčby obecně.
Objev tumor nekrotizujícího faktoru
Už v 90. letech 19. století zjistil newyorský chirurg William Coley, že akutní bakteriální infekce může způsobit regresi nádoru. V roce 1944 byl z Coleyho směsi bakteriálních toxinů izolován lipopolysacharid jako látka odpovědná za protinádorový účinek. Když byl injekčně podán myším s experimentálně vyvolaným nádorovým onemocněním, způsobil regresi nádoru [1].
V roce 1975 publikovali Carswell et al. experiment, v němž prokázali, že regrese nádoru po aplikaci endotoxinu ze Serratia marcescens myším je způsobena nikoli samotným toxinem, ale substancí nalezenou v séru experimentálních myší, kterou nazvali „tumor nekrotizující faktor“ [2]. V roce 1985 Beutler et al. studovali faktor, jenž způsoboval kachexii působením na lipoproteinovou lipázu a další metabolické dráhy. Později bylo zjištěno, že tento faktor, který nazvali „kachektin“, je totožný s tumor nekrotizujícím faktorem [3]. Současně Dayer et al. při hledání faktoru zprostředkovávajícího šok izolovali látku z buněk monocytární linie, u níž se také zjistilo, že je vlastně tumor nekrotizujícím faktorem [4]. Již tyto tři nezávislé objevy demonstrovaly klíčový význam tumor nekrotizujícího faktoru (TNF) v imunitním systému lidského organizmu.
Využití monoklonálních protilátek jako léčiv
Existence monoklonálních protilátek byla popsána v 70. letech 20. století u maligního myelomu ve formě paraproteinu. Jde o imunoglobuliny získané z klonální populace jediné plazmatické buňky, stejné chemické struktury a s monovalentní afinitou, tedy vážící se na přesně definované místo (epitop) antigenu. V roce 1975 G. Köhler a C. Milstein vytvořili fúzí myelomových buněčných linií s B-buňkami nesmrtelné hybridomy, které dokázaly produkovat specifické protilátky proti známým antigenům. Britský tým G. Wintera publikoval v roce 1988 svůj objev humanizace monoklonálních protilátek, který otevřel cestu k jejich terapeutickému použití v humánní medicíně [5].
Největší zásluhy o přenos technologie monoklonálních protilátek do farmakoterapie má zřejmě americký lékař L. M. Nadler, který jednak monoklonální protilátky použil ke klasifikaci lidských B-buněčných leukemií a lymfomů, ale také pracoval na vytvoření terapeutických monoklonálních protilátek a jako první na světě podal monoklonální protilátku pacientovi s B-lymfomem [6].
První monoklonální protilátkou zavedenou do klinické praxe jako léčivý přípravek byl muromonab-CD3, monoklonální protilátka proti membránovému CD3 receptoru T-lymfocytů. V USA byl schválen v roce 1986 pro léčbu akutních rejekčních reakcí po transplantaci ledviny, jater nebo srdce rezistentních na glukokortikoidy. Zajímavé je, že byl také vůbec prvním léčivým přípravkem, který byl registrován centralizovanou procedurou ve státech dnešní Evropské unie [7]. Poměrně dlouhou dobu však zůstal jediným zástupcem této nové skupiny léčiv.
Obrat přineslo v 90. letech zvládnutí technologie výroby chimérických protilátek produkovaných geny, jejichž sekvence DNA je ze 75 % lidská a pouze antigen vážící fragment (Fab) zůstává myší. První takovou protilátkou zavedenou do klinické praxe byl Fab fragment abciximab, registrovaný pro prevenci trombotických komplikací angioplastik, a první kompletní chimérická monoklonální protilátka rituximab byla schválena v roce 1997 pro léčbu lymfomů [8]. Všechny tehdy registrované monoklonální protilátky však byly určeny pouze pro krátkodobé podávání.
Počátky vývoje infliximabu
Chimérickou monoklonální protilátku proti TNF, pro kterou dnes používáme generický název infliximab, vyvinuli Junming Le a bratislavský rodák Ján Vilček pracující na New York University School of Medicine ve spolupráci s firmou Centocor (později Janssen Biotech) pod názvem cA2 [9]. Na základě experimentálních poznatků, podle nichž blokáda TNF zastavila rozvoj septického šoku u laboratorních zvířat, kterým byla podána letální dávka endotoxinu, byl pro klinické testování zvolen jako terapeutický cíl právě septický šok. Vzhledem k výsledkům předběžného klinického testování, v nichž nebyl prokázán přínos oproti placebu, byl však tento směr vývoje ukončen [10].
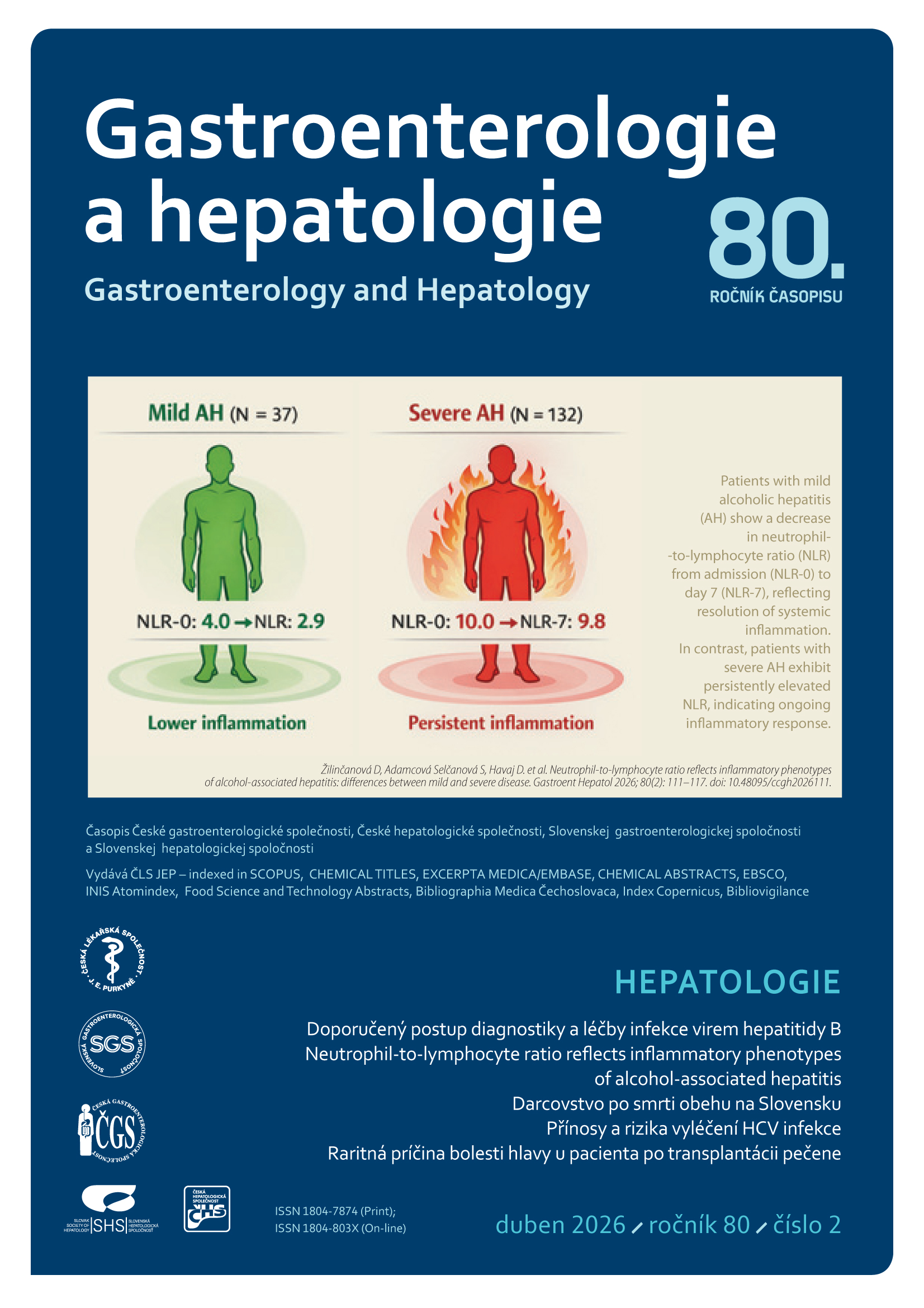
Závěr většiny odborníků v oboru byl, že inhibice jediného cytokinu není dostatečně účinná pro zastavení tak komplexního děje, jakým je zánětlivá reakce organizmu. Vzhledem k tomu byla očekávání spojená s dalším vývojem anti-TNF terapie u zánětlivých onemocnění nepříliš optimistická (obr. 1) [11].
Autoimunitní nemoci jako nový cíl
Od roku 1985 vedli M. Feldmann a R. Maini v Kennedyho institutu sídlícím tehdy v kampusu Charing Cross Hospital v Londýně výzkum role cytokinů u revmatoidní artritidy. Tato nemoc byla jako model autoimunitní choroby výhodná z hlediska relativně snadné dostupnosti bioptických vzorků, případně i větších částí tkání získaných perioperačně. K velkému překvapení zjistili, že ve všech získaných vzorcích byly přítomny všechny cytokiny, které tehdejší metody dokázaly identifikovat, což bylo v rozporu s původními představami o jejich krátkodobém působení. Dalším neočekávaným zjištěním získaným na tkáňových kulturách a potvrzeným v experimentu na laboratorních zvířatech bylo to, že blokáda jediného cytokinu – TNF pomocí monoklonální protilátky – vede k inhibici syntézy ostatních, především IL-1, IL-6 a GM-CSF. To vedlo k formulaci zcela nové hypotézy, že TNF je na samém vrcholu prozánětlivé cytokinové kaskády [12].
Protože žádná z britských farmaceutických společností neměla zájem o klinické testování této hypotézy, použili M. Feldmann a R. Maini pro první klinickou proof-of-concept studii právě monoklonální protilátku cA2 americké firmy Centocor, tedy infliximab. Tato první studie s deseti pacienty byla zahájena v květnu 1992. Po opakovaném podání dávky 20 mg/kg během 2 týdnů byla u všech zaznamenána pozitivní klinická i laboratorní odpověď [13]. Zveřejnění těchto poznatků vedlo k obratu v zájmu výrobců směrem k využití anti-TNF protilátek k léčbě revmatoidní artritidy, ale zejména v zájmu odborníků na jiné autoimunitní choroby k využití blokády TNF v léčbě i těchto onemocnění.
Už v této první studii se však ukázalo, že efekt blokády TNF monoklonální protilátkou je pouze dočasný – všichni pacienti relabovali do 18 týdnů po ukončení její aplikace. Další studie provedená v letech 1995–1996 na 101 pacientech s revmatoidní artritidou tedy musela zodpovědět řadu otázek, především zda je opakované podávání anti-TNF monoklonální protilátky bezpečné, zda imunogenicita nezruší její účinek a zda zánět nezačne probíhat alternativní cestou. Tato stude, již placebem kontrolovaná a dvojitě zaslepená, prokázala dobrou účinnost a dlouhodobost odpovědi zejména v kombinaci s metotrexátem [14].
Zavedení infliximabu do klinické praxe
Přestože první klinické poznatky byly získány u revmatoidní artritidy, jako první indikace pro komerční vývoj infliximabu byla zvolena Crohnova choroba, neboť závažnost jejího průběhu a omezené možnosti konvenční terapie nabízely relativně rychlejší cestu ke schválení regulačními autoritami.
Jako první použili protilátku cA2 u Crohnovy choroby S. van Deventer et al. v Amsterdamu, když podali dvě dávky 12leté dívce s nemocí neodpovídající na žádnou standardní terapii. Efekt zaznamenali již po první dávce a pacientka dosáhla remise, která však trvala pouze 3 měsíce [15]. Stejná skupina potom navázala otevřenou proof-of-concept studií s jednorázovým podáním dávky 10 nebo 20 mg/kg u deseti pacientů s nemocí refrakterní ke kortikoidům. Osm z nich dosáhlo klinicky i endoskopicky dokumentované remise, která trvala průměrně 4 měsíce [16].
Podkladem pro žádost o registraci monoklonální protilátky cA2 se staly dvě klinické studie zahrnující pacienty s Crohnovou nemocí (CN). Krátkodobá 12týdenní studie se 108 pacienty se středně těžkou až těžkou CN refrakterní ke standardní léčbě porovnávala dávky 5, 10 a 20 mg/kg s placebem. Ve 12. týdnu byla klinická odpověď dosažena u 41 % léčených, zatímco v placebové skupině byla pouze u 14 %. Pokud jde o remisi, byl poměr 33 % ku pouhým 4 % [17].
Hned druhá klinická studie byla zaměřena na fistulující formu nemoci, vůči které nebyla známa žádná účinná farmakoterapie. Bylo do ní zahrnuto 94 pacientů trpících nejméně 3 měsíce abdominálními či perianálními píštělemi komplikujícími CN. Pacienti byli léčeni podáním infliximabu v týdnech 0, 2 a 6 v dávce 5 nebo 10 mg/kg, nebo placebem. Primární endpoint, tedy 50% redukce píštělí, byl dosažen u 68 % pacientů s nižší dávkou a 56 % s vyšší dávkou oproti 26 % v placebové skupině. Navíc u 55 % a 38 % léčených došlo k úplnému zhojení píštělí oproti 13 % v placebové skupině. Medián doby, po kterou zůstaly píštěle uzavřené, dosáhl 3 měsíců [18].
Na základě těchto dvou studií americká Food and Drug Administration (FDA) schválila 24. srpna 1998 zrychlenou procedurou infliximab pod výrobním názvem Remicade pro léčbu CN. Schválení Evropskou lékovou agenturou (EMA) následovalo o rok později. Regulační autority však požadovaly provedení studií fáze III klinického hodnocení v udržovací léčbě.
Udržovací léčba
Odpověď na otázku, zda dlouhodobá blokáda TNF může vést k setrvalé léčebné odpovědi, se jako první pokusila nalézt studie ATTRACT. V této multicentrické, randomizované, placebem kontrolované studii fáze III bylo zařazeno 428 pacientů s aktivní revmatoidní artritidou, kteří poslední 3 měsíce užívali metotrexát. Po třech úvodních dávkách byli léčeni infliximabem i.v. každé 4 týdny nebo 8 týdnů v dávce 3 nebo 10 mg/kg. Ve 30. týdnu dosáhlo 20% zlepšení 50–58 % pacientů léčených různými dávkovacími schématy infliximabu oproti 20 % v kontrolní skupině. Celkem 50% zlepšení bylo dosaženo u 26–31 % léčených oproti pouhým 5 % s kombinací placebo a metotrexát [19].
U CN byla účinnost a bezpečnost udržovací terapie hodnocena ve studiích ACCENT I u luminální formy nemoci a ACCENT II u formy fistulující. V první studii bylo 573 pacientů randomizováno do tří skupin, ve kterých byli po úvodní dávce infliximabu léčeni buď placebem, nebo dávkami 5 mg/kg a 10 mg/kg i.v. à 8 týdnů. Odpověď na první dávku byla přítomna u 335 (58 %) pacientů, z nichž potom ve 30. týdnu studie bylo v remisi 21 % v placebové skupině a 39, resp. 45 % ve skupinách léčených infliximabem. Medián ztráty odpovědi byl 19 týdnů u placeba oproti 38 a a 54 týdnům v léčených skupinách [20].
Ve studii ACCENT II bylo zařazeno 306 pacientů s abdominálními nebo perianálními píštělemi, z nichž na úvodní tři dávky odpovědělo 195 osob. Ty byly dále randomizovány do skupiny s placebem nebo léčené dávkou 5 mg/kg à 8 týdnů. Jako primární endpoint byla hodnocena doba do ztráty odpovědi, která činila 14 týdnů u placebové skupiny oproti > 40 týdnům ve skupině léčené infliximabem. V 54. týdnu studie nemělo žádné píštěle 36 % léčených infliximabem oproti 19 % v placebové skupině [21].
Studie III. fáze klinického hodnocení tedy na klíčové otázky ohledně účinnosti a bezpečnosti dlouhodobé udržovací terapie odpověděly kladně. Navíc odhalily důležitý fakt dynamiky imunogenicity. Nejenže tvorba protilátek proti léčivu (ADA – anti-drug antibody) snižuje plazmatické koncentrace léčiva a jeho klinickou účinnost, ale i naopak nízké plazmatické koncentrace účinné látky vedou ke zvýšení tvorby těchto protilátek [11]. Tento fakt se později ukáže jako velmi významný.
Rozšíření indikací
Ke studiu účinnosti infliximabu u dalších autoimunitních onemocnění vedly kazuistiky zlepšení extraintestinálních manifestací CN, především kloubních a kožních, a také předpoklad podobné autoimunitní patofyziologie ulcerózní kolitidy (UC), psoriatické artritidy, ankylozující spondylitidy a psoriázy.
Jako první byla publikována studie německých autorů u ankylozující spondylitidy. Ve 12týdenní studii prokázali u 35 pacientů terapeutický efekt infliximabu v dávce 5 mg/kg ve srovnání s placebem [22]. Rozhodující pro schválení použití infliximabu v této indikaci byla studie ASSERT, ve které bylo primárním endpointem 20% zlepšení kritérií Assessment in Ankylosing Spondylitis (ASAS) International Working Group. Tohoto cíle dosáhlo ve 24. týdnu z celkem 279 pacientů 61 % léčených dávkou 5 mg/kg ve srovnání s 19 % v placebové skupině [23].
U UC byly provedeny dvě klíčové studie s názvem ACT 1 a 2 (Active Ulcerative Colitis Trials). V tomto programu bylo 364 pacientů s UC aktivní navzdory konvenční léčbě léčeno dávkami 5 nebo 10 mg/kg i.v. v týdnech 0, 2, 6 a potom à 8 týdnů, nebo placebem ve stejném režimu do 22. (ACT II) nebo 46. týdne (ACT I). Ve studii ACT I dosáhlo klinické odpovědi v 8. týdnu 69 % pacientů léčených dávkou 5 mg/kg a 61 % léčených 10 mg/kg ve srovnání s 37 % pacientů, kteří dostali placebo. Ve studii ACT II byly odpovídající hodnoty 64, 69 a 29 %. V 54. týdnu studie ACT I byla klinická odpověď přítomna u 45 a 44 % léčených oproti 20 % v placebové skupině [24].
Pro léčbu psoriatické artritidy byl infliximab testován v programu IMPACT. Ve studii III. fáze klinického hodnocení IMPACT 2 bylo 200 nemocných s aktivní psoriatickou artritidou neodpovídající na konvenční léčbu randomizováno do skupiny léčené infliximabem i.v. 5 mg/kg v týdnech 0, 2, 6, 14 a 22, nebo placebem. Primárním měřítkem klinické odpovědi bylo skóre ACR20 (American College of Rheumatology 20% Response). Klinická odpověď byla ve 14. týdnu studie dosažena u 58 % pacientů v léčené a 11 % v placebové skupině. Významný efekt infliximabu byl v této studii prokázán i na konkomitantní kožní projevy psoriázy, daktylitidu a entezopatie [25].
Na testování účinnosti a bezpečnosti infliximabu v léčbě samotné psoriázy byl zaměřen program studií EXPRESS. V multicentrické, randomizované a dvojitě slepé studii III. fáze klinického hodnocení bylo zařazeno 375 pacientů se středně těžkou až těžkou psoriázou a bylo léčeno infliximabem v i.v. dávce 5 mg/kg v obvyklém dávkovém schématu, nebo placebem. Primárním endpointem bylo 75% zlepšení v PASI skóre (Psoriasis Area and Severity Index) do 10. týdne. Tohoto cíle dosáhlo 80 % léčených a pouze 3 % z těch, kterým bylo podáváno placebo, 90% zlepšení tohoto parametru bylo prokázáno u 57 vs. 1 % pacientů v obou skupinách. V 50. týdnu sledování dosáhlo 61 % léčených skóre PASI75 a 45 % léčených dokonce PASI90 [26]. Studie v této indikaci byly ve srovnání s ostatními pozoruhodné výjimečně nízkým efektem placeba. O to impresivnější byl efekt inhibice TNF infliximabem.
Pro všechny výše uvedené indikace získal infliximab schválení regulačních autorit v letech 2003–2006. Další rozšíření registrovaných indikací následovalo v roce 2006 pro CN u dětí a v roce 2011 pro UC v petrické populaci [11]. Dá se říct, že ve všech těchto indikacích se léčba infliximabem postupně stala state-of-the-art terapií, se kterou jsou porovnávány všechny modernější léčebné postupy. V roce 2007 byl infliximab podán miliontému pacientovi, v roce 2013 již počet léčených přesáhl dva miliony [11]. I když byly do terapie zavedeny další látky blokující účinek TNF, žádná nepřekonala svou účinností ani bezpečností infliximab natolik, aby ho dokázala nahradit. A ani postupné zavádění jiných biologických léků s novými mechanizmy účinku zatím bohužel nepřineslo pokrok srovnatelný s nástupem první anti-TNF monoklonální protilátky. Přínos moderní medicíně vědců a lékařů, kteří se na tom podíleli, stejně jako firmy Centocor (později Janssen Biotech) je naprosto neocenitelný, a jak to často bývá, není příliš známý ani mezi odbornou, natožpak laickou veřejností.
Výzkum patofyziologie TNF
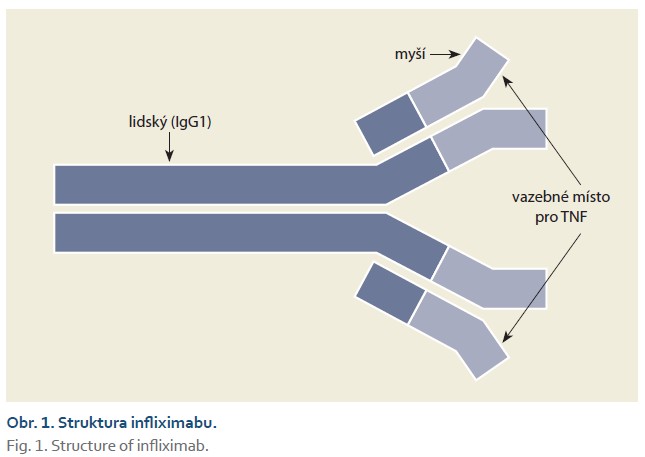
Zavedení farmakoterapie založené na inhibici funkce TNF do léčby autoimunitních onemocnění (obr. 2) pochopitelně urychlilo výzkum v oblasti cytokinů i samotného TNF. I stručné shrnutí současného stavu poznání o tomto mediátoru zánětu přesahuje možnosti tohoto přehledu, nicméně některé poznatky stojí za to zmínit.
V roce 1985 bylo zjištěno, že TNF má významnou sekvenční a funkční podobnost s již známým cytokinem lymfotoxinem. To vedlo k přejmenování tumor nekrotizujícího faktoru na TNFα a lymfotoxinu na TNFβ. V roce 1993 však byl objeven další cytokin velmi podobný lymfotoxinu, který byl nazván lymfotoxin-β. V roce 1998 byl proto na 7. mezinárodním kongresu o TNF přejmenován TNFβ na lymfotoxin-α, zatímco TNFα byl opět pojmenován pouze TNF [27].
Lidský gen TNF se nachází na chromozomu 6p21.3 v oblasti třídy III hlavního histokompatibilního komplexu, která obsahuje mnoho genů imunitního systému, přibližně 250 kilobází centromericky od lokusu HLA-B a 850 kilobází telomericky od HLA-DR. Sekvence v rámci 1100 párů bází DNA mezi 3’ koncem genu pro lymfotoxin-α a 5’ koncem genu TNF se ukázaly jako klíčové pro kontrolu transkripce [28].
TNF je rychle produkován v reakci na mnoho podnětů různými typy buněk. Největší množství produkují monocyty a makrofágy, ale významná je i produkce T- a B-lymfocyty. Dalšími buňkami, které exprimují TNF, jsou mastocyty, dendritické buňky a fibroblasty. Podněty, které aktivují gen TNF, zahrnují zevní stresory, patogenní látky a cytokiny z jiných imunitních buněk, zejména interleukin-1, interleukin-2, interferon-γ a samotný TNF. Transkripce TNF nezávisí na syntéze nových proteinů, což umožňuje rychlou aktivaci genu [29].
TNF je nejprve produkován jako transmembránový protein typu II (tmTNF), který je poté štěpen TNF konvertujícím enzymem (TACE) na rozpustnou formu (sTNF) a vylučován z buňky. Jednotlivé molekuly TNF jsou inertní, aktivní je až homotrimer vzniklý spojením tří molekul [30].
Účinek TNF na cílové buňky zprostředkovávají dva typy receptorů – TNFR1 a TNFR2. Volný TNF aktivuje pouze první typ, zatímco transmembránový může aktivovat oba typy receptorů, a navíc spouštět zánětlivé signální cesty své vlastní buňky [31]. Terapeuticky využívané anti-TNF látky neutralizují rozpustný TNF, ale vykazují rozdílné účinky na transmembránový TNF na různých buňkách, které ho exprimují. Tím lze vysvětlit určité rozdíly v jejich klinických účincích.
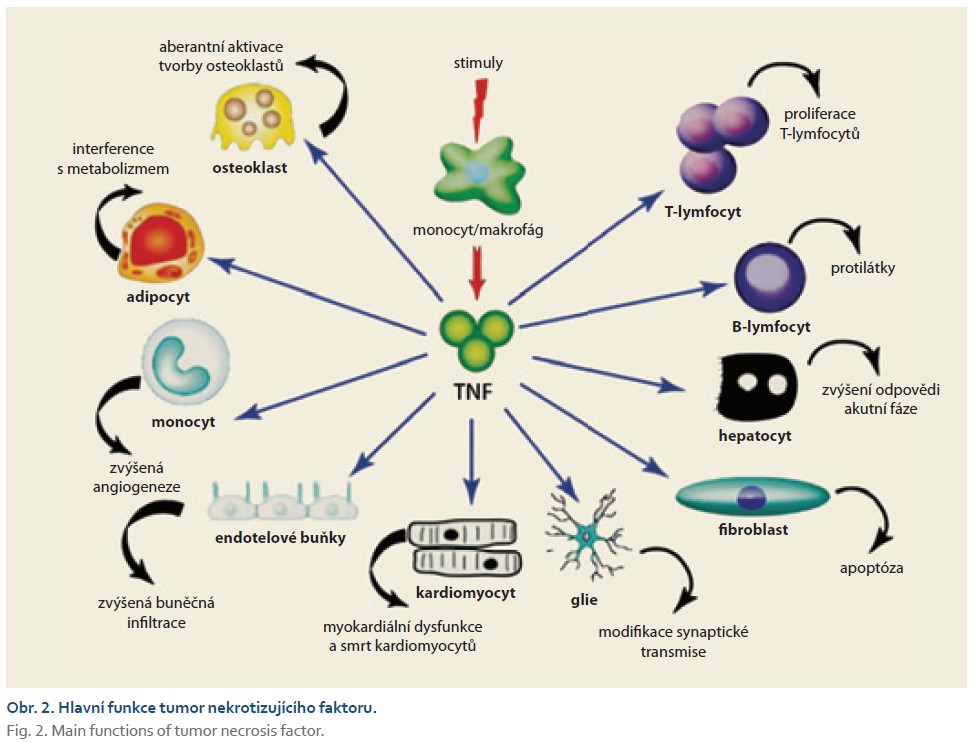
TNF hraje významnou roli i v patofyziologii dalších autoimunitních onemocnění. Infliximab je v léčbě některých z nich efektivní, ale k jejich léčbě schválen pouze v některých částech světa (jako u Kawasakiho a Behcetově nemoci v Japonsku), u některých jeho efektivita vedla k vývoji a schválení jiných anti-TNF léčiv (jako hidradenitis suppurativa nebo autoimunitní uveitida), u jiných byl pozitivní účinek v proof-of-concept studiích převážen rizikem nežádoucích účinků (jako SLE nebo autoimunitní hepatitida). Naopak v léčbě neautoimunitních chorob účinnost blokády TNF prokázána nebyla, byť v jejich patofyziologii hraje TNF jednu z hlavních rolí (obr. 3) [11].
Průkaz závislosti účinku na plazmatické koncentraci
Pokrok v terapii infiximabem přinesl také výzkum příčin jeho neúčinnosti, resp. selhání u některých pacientů. Jedním z vysvětlení je samozřejmě to, že zánět dokáže obejít blokádu TNF a probíhat dále alternativními cestami pomocí zvýšené aktivity jiných cytokinů. Alternativní cesta zánětu může být vysvětlením jak neúčinnosti blokády TNF u některých autoimunit, tak i primární absence odpovědi u 20–40 % pacientů napříč indikacemi a zčásti sekundární ztráty účinnosti u části pacientů, u kterých tento způsob léčby primárně zafungoval [11]. Nicméně významným důvodem může být i nedostatečná blokáda TNF způsobená poddávkováním, resp. přesněji nedosažením účinných koncentrací infliximabu v místě účinku. Příčinou může být nízká dávka, zvýšená clearance účinné látky i tvorba ADA. Příznivý efekt eskalace dávky při nedostatečné odpovědi byl prokázán již v roce 2007 u revmatoidní artritidy [32] a podobná pozorování byla k dispozici i u CN.
První důkazy pro závislost účinku infliximabu na minimální (údolní) plazmatické koncentraci přinesla analýza farmakokinetických dat získaných ve studii EXPRESS. V této studii pacienti, kteří měli před podáním udržovací dávky detekovatelné plazmatické koncentrace infliximabu, udrželi 75% zlepšení PASI skóre do konce studie významně pravděpodobněji než ti, u nichž byl infliximab před podáním nedetekovatelný, a to nezávisle na přítomnosti ADA [26].
Závislost účinku na minimální plazmatické koncentraci u CN byla prokázána cílenou studií kanadských autorů publikovanou v roce 2006. V této studii byly měřeny minimální plazmatické koncentrace a zjišťovány ADA u 105 pacientů, kteří byli po indukci dávkou 5 mg/kg léčeni buď udržovacími dávkami à 6–8 týdnů, nebo epizodickými reindukčními dávkami. I zde byla prokázána vyšší pravděpodobnost setrvalé remise u pacientů s detekovatelnými koncentracemi infliximabu před podáním další dávky nezávisle na přítomnosti ADA. Ti měli také nižší hodnoty CRP a vyšší podíl endoskopického zhojení. Současné podávání imunosupresiv nemělo na výsledek vliv [33].
Koncentrační závislost účinku anti-TNF monoklonálních protilátek byla potvrzena řadou dalších studií, v nichž byly také identifikovány klíčové faktory ovlivňující jejich farmakokinetiku. Nejvýznamnější je přítomnost ADA, která zvyšuje jejich clearance až trojnásobně. Slabší, ale významný dopad mají vysoké vstupní koncentrace TNF a CRP, hypoalbuminemie, vysoký body mass index (BMI) a mužské pohlaví. Clearance anti-TNF monoklonálních protilátek naopak snižuje současné podávání imunosupresiv [34].
Postupně získané poznatky vedly v roce 2017 k vydání doporučení American Gastroenterological Association u IBD monitorovat údolní plazmatické koncentrace infliximabu (a ostatních anti-TNF protilátek) a stanovení jeho doporučené minimální údolní plazmatické koncentrace na 5 mg/l [35]. Dosud zůstává doporučeno retroaktivní monitorování, tedy až při zjištění nedostatečné účinnosti. O možnosti proaktivního monitorování stejně jako o ideálních cílových hodnotách pro jednotlivé indikace se stále diskutuje, stejně jako o významu monitorování v jiných indikacích. Průkaz koncentrační závislosti účinku blokády TNF však nepochybně podstatně přispěl ke zvýšení významu subkutánních lékových forem.
Zavedení biosimilars do klinické praxe
Infliximab byl první monoklonální protilátkou, které vypršela v některých zemích EU patentová ochrana. Jako jeho první biologicky podobný léčivý přípravek (biosimilar) byl Evropskou lékovou agenturou (EMA) v roce 2014 registrován přípravek s označením CT-P13 (Remsima®), který vyvinula korejská firma Celltrion [36]. Díky tomuto vstupu konkurence na trh však došlo k razantnímu poklesu velmi vysokých cen biologických léčiv používaných v revmatologii, gastroenterologii a dermatologii. To umožnilo využít tuto vysoce účinnou léčbu i u pacientů, kteří by se k ní z ekonomických důvodů dříve nedostali.
Hlavním principem kategorie biologicky podobných léčiv je extrapolace indikací. Znamená to, že u biosimilar se provádí klinické hodnocení u omezeného počtu indikací (u infliximabu CT-P13 to byla ankylozujicí spondylitida ve studii PLANETAS a revmatoidní artritida ve studii PLANETRA), a na základě výsledků pak může být povolena registrace i pro léčbu jiných onemocnění, u kterých se předpokládá podobný mechanizmus terapeutického účinku. Registrace biosimilárního infliximabu CT-P13 byla první, u níž byl princip extrapolace použit pro léčivo charakteru monoklonální protilátky.
Přes shodné stanovisko všech regulačních autorit i odborníků nebyl princip extrapolace přijímán odbornou veřejností snadno. Teprve řada dat z klinické praxe o účinnosti a bezpečnosti přinesla zlom v jejich vnímání. Nejdůležitějším milníkem byla studie NORSWITCH publikovaná v roce 2017, která přinesla přesvědčivé důkazy o bezpečnosti a účinnosti záměny originálního infliximabu za biosimilární CT-P13 [37]. V současné době je dostupná řada biosimilárních přípravků anti-TNF monoklonálních protilátek a jejich používání je zcela standardní praxí.
Hlavním přínosem biosimilárních kopií originálních biologických léčiv je samozřejmě jejich nižší cena a konkurence mezi více přípravky, která náklady na ně dále snižuje. Tento zdánlivě pouze ekonomický přínos je však neocenitelný i z medicínského hlediska, neboť překonání ekonomických bariér umožní přístup k léčbě pacientům v méně bohatých regionech, kteří dříve čekali měsíce v pořadnících nebo k léčbě infliximabem neměli přístup vůbec. U infliximabu jde doslova o statisíce nemocných se závažnými autoimunitními nemocemi, kteří díky zavedení biosimilars mohou být léčeni, jakmile jsou k léčbě indikováni.
Subkutánní léková forma jako první biobetter
Jak již bylo uvedeno výše, u anti-TNF monoklonálních protilátek byla prokázána závislost účinku na minimální plazmatické koncentraci. Je-li tato koncentrace nedostatečná a nedostatečný je i klinický efekt léčby, lze dosáhnout zlepšení efektu zvýšením minimální plazmatické koncentrace. Právě s tímto cílem byl designován přípravek subkutánního infliximabu CT-P13.
Jako farmakokineticko-farmakodynamické cíle pro optimální dávkování byly stanovena dva parametry:
- dosažení minimálních plazmatických koncentrací nejméně 1 mg/l pro indikaci revmatoidní artritida a 5 mg/l pro ostatní indikace;
- dosažení celkové expozice infliximabu vyjádřené jako plocha pod křivkou plazmatických koncentrací (AUC) po dobu 8 týdnů v ustáleném stavu odpovídající dávce 3 mg/kg u revmatoidní artritidy a 5 mg/kg u ostatních indikací.
Pro splnění těchto požadavků bylo na základě sběru farmakokinetických dat v malé otevřené studii a na jejich základě provedeného populačního farmakokinetického modelování identifikováno jako optimální dávkovací schéma subkutánní podání 120 mg à 2 týdny. To bylo dále ověřeno v dose-finding částech registračních klinických studií [38].
Pro klinickou praxi je velmi významné i to, že uvedené cíle jsou dosahovány v širokém rozmezí hmotnosti pacientů bez nutnosti navyšovat dávkování subkutánního přípravku. Znamená to, že pro pacienty minimálně do hmotnosti 150 kg lze použít standardní dávku. Ve většině případů je farmakokinetických cílů dosaženo i u nemocných léčených velmi vysokým dávkováním intravenózně podávaného infliximabu při intenzifikované terapii.
Zlepšení farmakokinetických vlastností pomocí subkutánní aplikace vedlo navíc ke snížení imunogenicity infliximabu. Subkutánní forma infliximabu CT-P13 v klinickém hodnocení vykazuje konzistentně stejnou nebo nižší imunogenicitu, než má stejné léčivo ve formě intravenózní infuze. V registrační studii u pacientů s revmatoidní artritidou byl výskyt protilátek proti léčivu shodný v obou větvích, přičemž ve 22. týdnu byla imunogenicita subkutánní formy numericky nižší [39]. Tento jev je vysvětlován dosažením vysokozónové imunitní tolerance vysokými plazmatickými koncentracemi monoklonální protilátky.
Díky uvedeným vlastnostem splnil infliximab CT-P13 SC (Remsima® 120 mg firmy Celltrion) registrovaný v roce 2022 jako první léčivý přípravek monoklonální protilátky charakteristiky biobetter, tedy kopie, která není originálu pouze dostatečně podobná, ale je v některém z parametrů lepší než originální léčivý přípravek.
Nová intravenózní léková forma
I když zavedení subkutánní lékové formy přináší řadě pacientů i zdravotnickým zařízením významný benefit, neznamená to, že by i.v. aplikace infliximabu byla zastaralá či nepotřebná. Pro řadu pacientů je stále výhodnější i.v. aplikace à 8 týdnů než subkutánní à 2 týdny. Důvody mohou být různé, od nemožnosti dostatečně kvalitního zvládnutí aplikační techniky až po kožní onemocnění či potřeby rychlého nástupu účinku v úvodu terapie. Vzhledem k neustále narůstajícím počtům léčených však přetrvává problém přetíženosti ambulancí či stacionářů, ve kterých je biologická léčba aplikována.
Jednou z cest, jak snížit přetížení personálu, je usnadnění manipulace s lékovou formou nahrazením lyofilizátu určeného k rekonstituci přípravkem v roztoku, který může být přímo naředěn do infuze. Absence nutnosti rekonstituce navíc snižuje riziko kontaminace při její přípravě. Navíc je možné použití menších lahviček a balení, což snižuje náklady a prostorové nároky na skladování přípravku. V roce 2025 byla publikována práce, která se zabývala odhadem předpokládaného přínosu a úspor nákladů při používání takové lékové formy. Na základě dotazování odborníků došli autoři k závěru, že tekutá i.v. forma infliximabu může zkrátit čas přípravy aplikace o cca 50 % a náklady až o 20 %. Ještě zajímavější jsou odhadované finanční úspory pro západoevropské země, které se ročně pohybují ve stovkách tisíc eur [40].
Z těchto důvodů uvedla v roce 2026 firma Celltrion na trh svůj biosimilární infliximab CT-P13 ve formě koncentrátu pro infuzní roztok s koncentrací 40 mg/ml. Nová forma má stejnou účinnost a bezpečnostní profil jako prášek pro přípravu koncentrátu pro infuzní roztok a přináší několik praktických benefitů. Na rozdíl od původní lékové formy se již nerozpouští, ale přímo se ředí do infuzního vaku. To zkracuje čas přípravy o více než 50 % a snižuje riziko chyb při manipulaci. Navíc tato forma přináší úsporu místa v chladicích zařízeních díky kompaktnímu balení. Vzhledem k potřebě nižšího počtu lahviček je manipulace při přípravě infuze jednodušší, a snižuje se tak nejen čas na přípravu, ale i případné riziko kontaminace. Navíc je menší spotřeba jednorázového materiálu, jako jsou injekční stříkačky a jehly. Vzhledem k obsahu sorbitolu v roztoku je tato léková forma kontraindikována pro pacienty s hereditární intolerancí fruktózy, kterou je třeba před zahájením léčby vyloučit odebráním detailní anamnézy.
Nová léková forma je k dispozici v lahvičce s obsahem 100 mg nebo 350 mg infliximabu (40 mg/ml). Doba použitelnosti nové formy je před naředěním 4 roky při teplotě 2–8 °C a jednorázově může být uchovávána po dobu až 15 dnů při teplotách do 30 °C. Po naředění byla prokázána chemická a fyzikální stabilita po dobu až 60 dní při 2–8 °C a po vyjmutí z chladničky dalších 24 hod při 30 °C. I tato inovace farmaceutického charakteru může tedy zlepšit dostupnost a efektivitu léčby infliximabem v klinické praxi.
Závěr
Lze konstatovat, že infliximab je jedním z nevelkého množství skutečně převratných léčiv. Nejenže zásadním způsobem posunul možnosti farmakoterapie řady autoimunitních onemocnění, ale přispěl i ke studiu jejich patofyziologie a k pokroku biologické léčby obecně. Ani s nástupem novějších léčiv za více než čtvrtstoletí svého používání neztratil svoje postavení etalonu, se kterým je porovnávána účinnost a bezpečnost všech nových léků těchto nemocí. Jak je vidět z uvedeného přehledu, ani jeho vývoj a výzkum zdaleka nekončí.
ORCID autora
K. Urbánek 0000-0002-3461-1649.
Doručeno/Submitted: 3. 3. 2026
Přijato/Accepted: 9. 3. 2026
Korespondenční autor
doc. MUDr. Karel Urbánek, Ph.D.
Ústav farmakologie
LF UP a FN Olomouc
Hněvotínská 3
779 00 Olomouc
karel.urbanek@fnol.cz
Literatura
1. van Loo G, Bertrand MJM. Death by TNF: a road to inflammation. Nat Rev Immunol 2023; 23(5): 289–303. doi: 10.1038/s41577-022-00792-3.
2. Carswell EA, Old LJ, Kassel RL et al. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A 1975; 72(9): 3666–3670. doi: 10.1073/pnas.72.9.3666.
3. Beutler B, Greenwald D, Hulmes JD et al. Identity of tumour necrosis factor and the macrophage-secreted factor cachectin. Nature 1985; 316(6028): 552–554. doi: 10.1038/316552a0.
4. Dayer JM, Beutler B, Cerami A. Cachectin/ tumor necrosis factor stimulates collagenase and prostaglandin E2 production by human synovial cells and dermal fibroblasts. J Exp Med 1985; 162(6): 2163–2168. doi: 10.1084/jem.162.6.2163.
5. Riechmann L, Clark M, Waldmann H et al. Reshaping human antibodies for therapy. Nature 1988; 332(6162): 323–327. doi: 10.1038/332 323a0.
6. Nadler LM, Roberts WC. Lee Marshall Nadler, MD: a conversation with the editor. Proc (Bayl Univ Med Cent) 2007; 20(4): 381–389. doi: 10.1080/08998280.2007.11928327.
7. Smith SL. Ten years of Orthoclone OKT3 (muromonab-CD3): a review. J Transpl Coord 1996; 6(3): 109–119. doi: 10.7182/prtr.1.6.3.8145l3u185493182.
8. Reichert JM. Monoclonal antibodies in the clinic. Nat Biotechnol 2001; 19(9): 819–822. doi: 10.1038/nbt0901-819.
9. Knight DM, Trinh H, Le J et al. Construction and initial characterization of a mouse-human chimeric anti-TNF antibody. Mol Immunol 1993; 30(16): 1443–1453. doi: 10.1016/0161-5890(93)90106-l.
10. Vilcek J, Feldmann M. Historical review: cytokines as therapeutics and targets of therapeutics. Trends Pharmacol Sci 2004; 25(4): 201–209. doi: 10.1016/j.tips.2004.02.011.
11. Melsheimer R, Geldhof A, Apaolaza I et al. Remicade (infliximab): 20 years of contributions to science and medicine. Biologics 2019; 13: 139–178. doi: 10.2147/BTT.S 207246.
12. Monaco C, Nanchahal J, Taylor P et al. Anti-TNF therapy: past, present and future. Int Immunol 2015; 27(1): 55–62. doi: 10.1093/intimm/dxu102.
13. Elliott MJ, Maini RN, Feldmann M et al. Treatment of rheumatoid arthritis with chimeric monoclonal antibodies to tumor necrosis factor alpha. Arthritis Rheum 1993; 36(12): 1681–1690. doi: 10.1002/art.1780361206.
14. Maini RN, Breedveld FC, Kalden JR et al. Therapeutic efficacy of multiple intravenous infusions of anti-tumor necrosis factor alpha monoclonal antibody combined with low-dose weekly methotrexate in rheumatoid arthritis. Arthritis Rheum 1998; 41(9): 1552–1563. doi: 10.1002/1529-0131(199809) 41: 9<1552:: AID-ART5>3.0.CO; 2-W.
15. Derkx B, Taminiau J, Radema S et al. Tumour-necrosis-factor antibody treatment in Crohn‘s disease. Lancet 1993; 342(8864): 173–174. doi: 10.1016/0140-6736(93)91375-v.
16. van Dullemen HM, van Deventer SJ, Hommes DW et al. Treatment of Crohn‘s disease with anti-tumor necrosis factor chimeric monoclonal antibody (cA2). Gastroenterology 1995; 109(1): 129–135. doi: 10.1016/0016-5085(95)90277-5.
17. Targan SR, Hanauer SB, van Deventer SJ et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn‘s disease. Crohn‘s disease cA2 study group. N Engl J Med 1997; 337(15): 1029–1035. doi: 10.1056/NEJM199710093371502.
18. Present DH, Rutgeerts P, Targan S et al. Infliximab for the treatment of fistulas in patients with Crohn‘s disease. N Engl J Med 1999; 340(18): 1398–1405. doi: 10.1056/NEJM199905063401804.
19. Maini R, St Clair EW, Breedveld F et al. Infliximab (chimeric anti-tumour necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised phase III trial. ATTRACT Study Group. Lancet 1999; 354(9194): 1932–1939. doi: 10.1016/s0140-6736(99)05246-0.
20. Hanauer SB, Feagan BG, Lichtenstein GR et al. Maintenance infliximab for Crohn‘s disease: the ACCENT I randomised trial. Lancet 2002; 359(9317): 1541–1549. doi: 10.1016/S0140-6736(02)08512-4.
21. Sands BE, Anderson FH, Bernstein CN et al. Infliximab maintenance therapy for fistulizing Crohn‘s disease. N Engl J Med 2004; 350(9): 876–885. doi: 10.1056/NEJMoa030815.
22. Braun J, Brandt J, Listing J et al. Treatment of active ankylosing spondylitis with infliximab: a randomised controlled multicentre trial. Lancet 2002; 359(9313): 1187–1193. doi: 10.1016/s0140-6736(02)08215-6.
23. van der Heijde D, Dijkmans B, Geusens P et al. Efficacy and safety of infliximab in patients with ankylosing spondylitis: results of a randomized, placebo-controlled trial (ASSERT). Arthritis Rheum 2005; 52(2): 582–591. doi: 10.1002/art.20852.
24. Rutgeerts P, Sandborn WJ, Feagan BG et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med 2005; 353(23): 2462–2476. doi: 10.1056/NEJMoa050516.
25. Antoni C, Krueger GG, de Vlam K et al. Infliximab improves signs and symptoms of psoriatic arthritis: results of the IMPACT 2 trial. Ann Rheum Dis 2005; 64(8): 1150–1157. doi: 10.1136/ard.2004.032268.
26. Reich K, Nestle FO, Papp K et al. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: a phase III, multicentre, double-blind trial. Lancet 2005; 366(9494): 1367–1374. doi: 10.1016/S0140-6736(05)67566-6.
27. Grimstad Ø. Tumor necrosis factor and the tenacious α. JAMA Dermatol 2016; 152(5): 557. doi: 10.1001/jamadermatol.2015.4322.
28. Papadakis KA, Targan SR. Tumor necrosis factor: biology and therapeutic inhibitors. Gastroenterology 2000; 119(4): 1148–1157. doi: 10.1053/gast.2000.18160.
29. Falvo JV, Tsytsykova AV, Goldfeld AE. Transcriptional control of the TNF gene. Curr Dir Autoimmun 2010; 11: 27–60. doi: 10.1159/000289196.
30. Horiuchi T, Mitoma H, Harashima S et al. Transmembrane TNF-alpha: structure, function and interaction with anti-TNF agents. Rheumatology (Oxford) 2010; 49(7): 1215–1228. doi: 10.1093/rheumatology/keq031.
31. van Loo G, Bertrand MJM. Death by TNF: a road to inflammation. Nat Rev Immunol 2023; 23(5): 289–303. doi: 10.1038/s41577-022-00792-3.
32. Rahman MU, Strusberg I, Geusens P et al. Double-blinded infliximab dose escalation in patients with rheumatoid arthritis. Ann Rheum Dis 2007; 66(9): 1233–1238. doi: 10.1136/ard.2006.065995.
33. Maser EA, Villela R, Silverberg MS et al. Association of trough serum infliximab to clinical outcome after scheduled maintenance treatment for Crohn‘s disease. Clin Gastroenterol Hepatol 2006; 4(10): 1248–1254. doi: 10.1016/j.cgh.2006.06.025.
34. Ordás I, Mould DR, Feagan BG et al. Anti-TNF monoclonal antibodies in inflammatory bowel disease: pharmacokinetics-based dosing paradigms. Clin Pharmacol Ther 2012; 91(4): 635–646. doi: 10.1038/clpt.2011.328.
35. Feuerstein JD, Nguyen GC, Kupfer SS et al. American Gastroenterological Association Institute Guideline on therapeutic drug monitoring in inflammatory bowel disease. Gastroenterology 2017; 153(3): 827–834. doi: 10.1053/j.gastro.2017.07.032.
36. Urbánek K. První biosimilární monoklonální protilátka – infliximab. Klin Farmakol Farm 2014; 28(1): 19–22.
37. Jørgensen KK, Olsen IC, Goll GL et al. Switching from originator infliximab to biosimilar CT-P13 compared with maintained treatment with originator infliximab (NOR-SWITCH): a 52-week, randomised, double-blind, non-inferiority trial. Lancet 2017; 389(10086): 2304–2316. doi: 10.1016/S0140-6736(17)0068-5.
38. Iannone F, Conti F, Cauli A et al. Subcutaneously-administered infliximab in the management of rheumatoid arthritis: a short narrative review of current clinical evidence. J Inflamm Res 2022; 15: 3259–3267. doi: 10.2147/JIR.240593.
39. Westhovens R, Wiland P, Zawadzki M et al. Efficacy, pharmacokinetics and safety of subcutaneous versus intravenous CT-P13 in rheumatoid arthritis: a randomized phase I/III trial. Rheumatology (Oxford) 2021; 60(5): 2277–2287. doi: 10.1093/rheumatology/keaa580.
40. Nam K, Kwon TS, Di Biasio F et al. Perceived benefits and cost savings of liquid formulation of intravenous infliximab: perspectives of seven European countries. Expert Opin Biol Ther 2025; 25(9): 1017–1024. doi: 10.1080/14712598.2025.2544756.